Prof.Dr. Haydar BAĞIŞTıbbi Genetik ABD Başkanı 19 Ocak2020 FMF GenetiğiFMF, otozomal resesif kalıtım göstermektedir. Hasta bireyin anne ve babası zorunlu taşıyıcıdır. FMF taşıyıcılığı ve akraba evliliği oranı yüksek olan toplumlarda çocukların taşıyıcı veya hasta olarak dünyaya gelme olasılığı yüksektir. Eğer anne ve baba mutasyonu heterozigot olarak taşıyorsa bir sonraki nesilde hasta bireyin dünyaya gelme ihtimali %25, taşıyıcılık oranı %50 iken sağlıklı birey olma olasılığı %25’dir. Ailesel Akdeniz Ateşi, hekimlerin periyodik karın ağrısı, göğüs ağrısı, ateş ve artrit şikâyetleri ile tanıda zorlandığı bir hastalıktır. FMF ile benzer semptomlar gösteren hastalıkların ayırıcı tanısının güçlüğü göz önüne alındığında, moleküler tanı metotları önem kazanmıştır. Tüm genetik hastalıklarda olduğu gibi, FMF için de genetik tanı uygulaması yapılmalı ve danışma hizmeti verilmelidir. Her hastanın taşıdığı mutasyon tespit edilerek “genetik kimlik kartı” alması sağlanmalıdır. Hasta oranının 1:1000 ve taşıyıcı oranın 1:5 olduğu ülkemizde, FMF kaynaklı ciddi bir sağlık sorunu bulunmaktadır.Hastalarımızın FMF’in genetik boyutu hakkında ciddi şekilde bilgilendirilmesi ile sonraki jenerasyonlarda maddi-manevi kayıpların azaltılması hedeflenmelidir. SıklığıHastalığın ülkemizde görülme sıklığı 1/1000 iken, taşıyıcı sıklığı 1/5 gibi oldukça yüksek bir orandadır. Bu durum, özellikle akraba evliliklerinin çok sık olduğu ülkemiz için ayrı bir öneme sahiptir. Akdeniz çevresindeki ırklar ve etnik gruplarda (Sefardik Museviler, Ermeniler, Türkler ve Araplar) nispeten sık görülmektedir (3). FMF’den sorumlu olan MEFV geni 10 ekzondan oluşmaktadır. 2. ve 10. ekzonlar mutasyonların en fazla meydana geldiği ekzonlar olup, diğer ekzonlarda da mutasyonlar oluşabilmektedir. FMF’li olgularda en sık görülen M694V mutasyonu MEFV geninde 2080. nükleotitte (694. aminoasit) valinin metiyonin ile yer değiştirmesi sonucu oluşmaktadır. M694V homozigot mutasyonunu taşıyan FMF hastalarında kronik böbrek yetmezliği ile sonuçlanan amiloidoza gidiş oranı %55 olarak bildirilmiştir. Tanı KriterleriAilevi Akdeniz Ateşi tanısı konmasında klinik bulgular ve aile öyküsü ile biyokimyasal ve genetik laboratuvar verileri kullanılmaktadır.
KALITIMI
- Otozomal resesif
KARDİYOVASKÜLER
Kalp
- Perikardit
SOLUNUM
Akciğer
- Plörit
KARIN
Karaciğer
- Hepatomegali
Dalak
- Splenomegali
Gastrointestinal
- Peritonit - Karın ağrısı
Genitoüriner
Dış Cinsel Organ (Erkek)
- Tunica vajinalis iltihabı (orşit)
Böbrekler
- Nefrotik sendrom - Renal amiloidoz - Böbrek yetmezliği
İSKELET
- Monartiküler veya oligoartiküler artrit - Artralji
CİLT, TIRNAKLAR VE SAÇ
Deri
- Alt bacak ve ayak bileğinde geçici ağrılı erizipel benzeri lezyonlar
NÖROLOJİK
Merkezi sinir sistemi
- Menenjit
METABOLİK ÖZELLİKLER
- Ateş , epizodik
LABORATUVAR ANORMALLİKLERİ
- Belirgin lökositoz (30.000/ml) - Yüksek eritrosit sedimantasyon hızı
ÇEŞİTLİ
- Arap, Türk, Ermeni ve Sefarad Yahudi popülasyonlarında
yaygın - Başlangıç sıklıkla çocukluk veya ergenlik döneminde başlar - 24-48 saat süren akut ataklar - Atak sıklığı haftada birkaç kez ila yılda bir kez olabilir - Ayrıca bakınız otozomal dominant FMF ( 134610 ), MEFV genindeki heterozigot mutasyonların neden olduğu MOLEKÜLER TEMEL
- Pirin genindeki mutasyonlardan kaynaklanır (MEFV, 608107.0001 )
Kaynak: OMIM
FMF’in klinik tablosu, abdominal ağrı (peritonit) ve/veya plöritik ağrı ve/veya artrit (ayak bileği ve diz) ile birlikte, 12-96 saat sürebilen tekrarlayan ateş atakları ile karakterizedir. Hastalık belirtilerinin belirli periyotlarla ortaya çıkması tanı için en önemli kriterdir. Hasta eğer atak sırasında görülmüşse, atağa eşlik eden inflamatuvar bulgularının varlığı (lökositoz, sedimantasyon artışı, fibrinojen ve CRP’nin yükselmesi) ve bu testlerin atak sonlanınca normal değerlere inmesi tanıya yardımcı olur. Bu testlerin pozitif bulunmasının FMF’e özgü olmadığı, sadece vücutta inflamasyonun varlığına işaret ettiği unutulmamalıdır. Klinik pratikte, hastalığın tanısı için en sık Tel-Hashomer kriterleri kullanılmaktadır(4).
Aile öyküsünün pozitif olması tanıyı destekleyen önemli bir bulgudur. Bununla beraber, FMF’in resesif geçişli bir genetik hastalık olması nedeniyle, ailede başka bir indeks vaka bulunmayabileceği de unutulmamalıdır. Hastalığın Kalıtım Modeli ve Moleküler Genetiği FMF otozomal resesif geçişli bir hastalıktır, ancak literatürlerde az sayıda otozomal dominant geçişli vakalar da bildirilmiştir. FMF’den sorumlu olan MEFV geni, kromozom 16p13.3’de lokalizedir, 10 ekzondan oluşur ve Pyrin proteinini kodlar (Tablo 1). Tablo 1. FMF Genkart
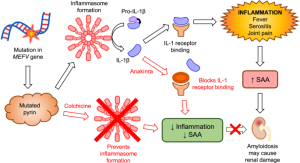
Pyrin proteini, başlıca nötrofil ve monositlerde sentezlenir ve kaspaz-1 ile interlökin-1β aracılığıyla apoptozda görev alacak proteinlerin ekspresyonunu ve anti-inflamatuvar aktivitelerini düzenler.
Hastalığa neden olan mutasyonlar, en fazla genin 2. ve 10. ekzonlarında bulunur. MEFV (MEditerranean FeVer); 10 ekzon içeren, 15 kilobazlık, 3505 nükleotidden oluşan bir gendir. Öncelikle 10. ekzonda hastalıkla ilgili üç yanlış anlamlı mutasyon (M694V, M680I, V726A) tanımlanmıştır. M694V MEFV geni 2080. nükleotidde, proteinde Metiyonin Valin aminoasit değişikliğiyle sonlanan, A>G transisyonu tanımlanmıştıR. MEFV geni cDNA dizisinde 1170. nükleotidde A>G değişimi gösterilmiştir. Türk toplumunda yaklaşık olarak %43.5 oranındadır. M680I MEFV geni 2040. nükleotidde, G>C transisyonu, ilk olarak Ermeni bir FMF hastada tanımlanmıştır (The International FMF Consortium 1997). Proteinde Metiyonin İzolösin aminoasit değişikliğiyle dizisinde nükleotid 1130’da G>C değişimini göstermiştir. Türk toplumunda görülme sıklığı %12 bulunmuştur. Yaygın olarak görülen dört MEFV mutasyonunun, Fenotip I (M694V %38, M680I %8, V726A %4, E148Q %4) ve Fenotip II (M694V %51.5, M680I %9, V726A %2.9, E148Q %3.5)
FMF araştırmasında klinik değerlendirmeyi takiben moleküler genetik incelemenin yapılması çok önemlidir. FMF tanısının gecikmesi Özellikle homozigot olmak üzere M694V mutasyonunu taşıyan Kuzey Afrikalı Yahudilerde %90 olguda mutasyonla amiloidoz gelişmesi arasında sıkı bir ilişki gösterilmiştir. Bazı çalışmalarda M694V mutasyonu ile ağır bir fenotip bildirilmesine rağmen, bu bulguyu desteklemeyen çalışmalar da mevcuttur(7-10). Bununla birlikte aile içi ve aileler arası klinik farklılıklara dayanarak fenotipin, MEFV lokusu dışındaki genler ve/veya çevresel etmenlerden etkilendiği düşünülmektedir. TedaviFMF önlenebilir ve kontrol edilebilir bir hastalıktır. Hastalığın oluşturduğu klinik tabloyu düzeltme amacıyla çiğdem bitkisinden elde edilen kolşisin kullanılmaktadır. Kolşisin, FMF tedavisinde 2 önemli amaçla kullanılır. 1. Atakların engellenmesi veya hafifletilmesi Düzenli olarak kolşisin kullanan hastalarda ataklar ya hiç tekrarlamaz ya da öncekilere oranla çok daha seyrek hale gelir ve hafif geçerler. Sadece atak döneminde kullanılmasının bir yararı yoktur ve bu şekilde başlamış olan atağa önleyici bir etki sağlamaz. Etkinliği ilacın düzenli kullanımına bağlıdır. 2. Amiloidoz gelişiminin engellenmesi Kolşisin, hekim kontrolünde düzenli ve yeterli dozda kullanıldığında amiloidoz gelişimini engellemektedir. M694V mutasyonunu taşıyan kişilerin amiloidoz gelişimi için daha yüksek riske sahip oldukları ve bu nedenle mutlaka kolşisin kullanımı gerektiği bildirilmektedir. Tedavi takibinde gerekli hassasiyetin gösterilmemesi durumlarında klinik tablo tekrar bozulabilmektedir. Sık Sorulan Sorular1. Ataklar FMF’de nasıl seyreder? FMF atakları tekrarlayan ateş ve ağrılı durumlardır ve önceden kestirilmesi zordur. Ataklar genelde 2-3 gün sürer ve en şiddetli atak ilk 12-24 saatte görülür.
4. c.2080A>G (p.Met694Val): Bu heterozigotların çoğunda hastalığın ana bulgusu peritonit gelişmeyen karın ağrısı ataklarıdır. Heterozigot mutasyon taşıyanlarda klnik önemlidir. Klinik bulgu varsa doktorunun istemiyle kolşisin kullanabilir. Referanslar:1. Shinar Y, Livneh A, Langevitz P, Zaks N, Aksentijevich I, Koziol DE, Kastner DL, Pras M, Pras E.Genotype-phenotype assessment of common genotypes among patients with familial Mediterranean fever. . J Rheumatol. 2000;27(7):1703-7.
|