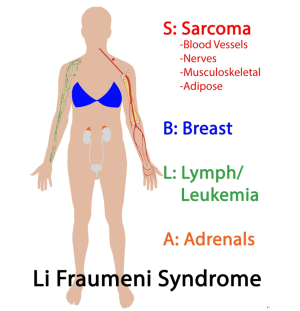
Li-Fraumeni sendromu (LFS), çok çeşitli çocukluk ve yetişkinlik başlangıçlı maligniteler için yüksek risklerle ilişkili bir kansere yatkınlık sendromudur. LFS'li bireylerde yaşam boyu kanser riski erkekler için ≥% 70 ve kadınlar için ≥% 90'dır. Beş kanser türü, LFS tümörlerinin çoğunu oluşturur: adrenokortikal karsinomlar, meme kanseri, merkezi sinir sistemi tümörleri, osteosarkomlar ve yumuşak doku sarkomları.
LFS, lösemi, lenfoma, gastrointestinal kanserler, baş ve boyun kanserleri, böbrek, gırtlak, akciğer, deri (örn. Melanom), yumurtalık, pankreas, prostat, testis ve tiroid dahil olmak üzere birçok ek kanser riskinde artış ile ilişkilidir. LFS'li bireyler, çocuklukta ve genç yetişkinlikte kanser için yüksek risk altındadır; Hayatta kalanlar, birden fazla birincil kanser için yüksek risk altındadır. Li-Fraumeni sendromlu kişilerde diğer bazı kanser türleri de daha sık görülür .
Li-Fraumeni sendromunun dünya çapında 5.000 ila 20.000 kişide 1’e ortaya çıktığı düşünülmektedir.
Li-Fraumeni sendromu , TP53 genindeki mutasyonlarla ilişkilidir . Li-Fraumeni sendromlu ailelerin yaklaşık dörtte üçünde TP53 geninde germ hattı mutasyonları vardır.
Germline mutasyonları tipik olarak kalıtsaldır ve vücuttaki her hücrede TP53 genin bir kopyasında mutasyon mevcuttur.
TP53 tümör baskılayıcı bir gendir, yani normalde hücrelerin büyümesini ve bölünmesini kontrol etmeye yardımcı olur.
Bu gendeki mutasyonlar, hücrelerin kontrolsüz bir şekilde bölünmesine ve tümör oluşturmasına izin verebilir. TP53 mutasyonları olan kişilerde diğer genetik ve çevresel faktörlerin de kanser riskini etkilemesi muhtemel gözükmektedir.
Li-Fraumeni sendromuna ve Li-Fraumeni benzeri sendroma özgü kansere sahip birkaç ailede TP53 mutasyonları yoktur. Bu vakalarda yer alan genetik faktörler belirsizdir.
Li-Fraumeni sendromu , otozomal dominant bir modelde kalıtılır; bu, her hücrede değiştirilmiş genin bir kopyasının kanser geliştirme riskini artırmak için yeterli olduğu anlamına gelir.
Li-Fraumeni sendromlu çoğu insan, etkilenen bir ebeveynden genin değiştirilmiş bir kopyasını miras alır.
Vakaların 7 ila 20 oranında ise, değişmiş gen bir sonucudur yeni (de novo) mutasyonu geni üreme hücrelerinin (yumurta veya sperm) oluşumu sırasında veya gelişimin çok erken döneminde meydana gelir.
Li-Fraumeni sendromunda bir kanserin gelişmesi için, bir kişinin yaşamı boyunca vücut hücrelerinde TP53 geninin diğer kopyasını içeren bir mutasyon meydana gelmelidir. Bu genin iki değiştirilmiş kopyasına sahip hücreler, tümörlerin gelişmesine izin veren fonksiyonel TP53 proteini yapmaz.
Bir TP53 gen mutasyonunu miras alan hemen hemen herkes, sonunda bazı hücrelerde genin ikinci kopyasında bir mutasyon kazanacaktır (çift vuruş hipotezi). İkinci mutasyon genellikle meme, kemik veya kas dokusundaki hücrelerde meydana gelir ve tipik olarak Li-Fraumeni sendromunda yaygın olan tümörlere yol açar.
TP53 geni (kromozom 17 p13.1), tümör proteini, p53 (veya p53) olarak adlandırılan bir proteinin yapmak için talimatlar içerir. Bu protein, bir tümör baskılayıcı görevi görür; bu hücrelerin çok hızlı veya kontrolsüz bir şekilde büyümesini ve bölünmesini (çoğalmasını) önleyerek hücre bölünmesini düzenlediği anlamına gelir. P53 proteini, DNA onarımını ve hücre bölünmesini düzenlemek için gerekli olduğundan, " TP53 geni genomun koruyucusu” yani genomun gardiyanı gibi işlev görür.
P53 proteini, doğrudan DNA'ya bağlandığı (bağlandığı) vücuttaki hücrelerin çekirdeğinde bulunur. Bir hücredeki DNA, güneş ışığından kaynaklanan toksik kimyasallar, radyasyon veya ultraviyole (UV) ışınları gibi maddeler tarafından hasar gördüğünde, bu protein, DNA'nın onarılıp onarılmayacağını veya hasarlı hücrenin kendi kendini yok edip etmeyeceğini belirlemede kritik bir rol oynar apoptoz). DNA onarılabilirse, p53 hasarı düzeltmek için diğer genleri etkinleştirir. DNA onarılamazsa, bu protein hücrenin bölünmesini önler ve apoptoza girmesi için sinyal verir. p53 mutasyona uğramış veya hasar görmüş DNA'ya sahip hücrelerin bölünmesini durdurarak tümör gelişimini önlemeye yardımcı olur.
Genetik Danışmanlık.
LFS, otozomal dominant bir şekilde miras alınır. LFS teşhisi konan çoğu birey, bir ebeveynden TP53 patojenik bir varyantı miras almıştır. De novo germline TP53 patojenik varyantına sahip bireylerin oranının % 7 ile % 20 arasında olduğu tahmin edilmektedir. LFS tanısı alan bir bireyin (yani, klasik LFS kriterlerini karşılayan ve / veya heterozigot germ hattı TP53 patojenik varyantına sahip bir birey).
LFS'ye neden olan patojenik bir varyantı kalıtım yoluyla alma ve ilişkili kanser risklerine sahip olma riski% 50'dir. LFS bakımından risk altındaki aile üyeleri için öngörücü testler , doğum öncesi testler ve ailede bir TP53 germ hattı patojenik varyantı tanımlanmışsa implantasyon öncesi genetik test mümkündür .
Li-Fraumeni benzeri sendromda ise, 22q12 kromozomundaki CHEK2(checkpoınt kinase 2) genindeki heterozigot mutasyona mutlaka bakılmasında fayda vardır.
Aşağıdaki maddelerden birisinin varlığı TP53 testi için endikasyon oluşturur
1) Ailede TP53 mutasyonu olan birey varlığı.
2) Li-Fraumeni kriteri olan kombinasyonun varlığı a) 45 yaş altında sarkom tanısı almış olmak b) En az bir birinci derece yakın akrabanın 45 yaş altında kanser tanısı alması c) Buna ek olarak ailenin aynı tarafından bir 1. ya da 2. derece yakın akrabanın herhangi bir yaşta sarkom tanısı alması ya da 45 yaş altında kanser tanısı alması.
3) Li-Fraumeni benzeri sendrom kriteri olan kombinasyonun varlığı a) 45 yaş altında çocukluk çağı sarkomları, beyin tümörü, adrenokortikal karsinom tanısı almış olmak b) En az bir birinci ya da ikinci derece yakın akrabanın herhangi bir yaşta tipik Li-Fraumeni tümörü tanısı alması c) Buna ek olarak ailenin aynı tarafından bir 1. ya da 2. derece yakın akrabanın 60 yaş altında kanser tanısı alması
4) 30 yaş altında meme kanseri tanısı aldığı halde BRCA1/BRCA2 mutasyonu negatif çıkan ve özellikle aile öyküsünde sarkom, beyin tümörü veya adrenokortikal karsinom olan hastalar.
25 Eylül 2020
Prof. Dr. Haydar BAĞIŞ
Kaynaklar.
Loyo M, Li RJ, Bettegowda C, Pickering CR, Frederick MJ, Myers JN, Agrawal N. Lessons learned from next-generation sequencing in head and neck cancer. Head Neck. 2013 Mar;35(3):454-63. doi: 10.1002/hed.23100. Epub 2012 Aug 21. Review.
Batalini F, Peacock EG, Stobie L, Robertson A, Garber J, Weitzel JN, Tung NM. Li-Fraumeni syndrome: not a straightforward diagnosis anymore-the interpretation of pathogenic variants of low allele frequency and the differences between germline PVs, mosaicism, and clonal hematopoiesis. Breast Cancer Res. 2019 Sep 18;21(1):107. doi: 10.1186/s13058-019-1193-1. Review.
Gargallo P, Yáñez Y, Segura V, Juan A, Torres B, Balaguer J, Oltra S, Castel V, Cañete A. Li-Fraumeni syndrome heterogeneity. Clin Transl Oncol. 2019 Nov 5. doi: 10.1007/s12094-019-02236-2. [Epub ahead of print] Review.
https://www.ncbi.nlm.nih.gov/books/NBK131.
Kratz CP, Achatz MI, Brugières L, Frebourg T, Garber JE, Greer MC, Hansford JR, Janeway KA, Kohlmann WK, McGee R, Mullighan CG, Onel K, Pajtler KW, Pfister SM, Savage SA, Schiffman JD, Schneider KA, Strong LC, Evans DGR, Wasserman JD, Villani A, Malkin D. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res. 2017 Jun 1;23(11):e38-e45. doi: 10.1158/1078-0432.CCR-17-0408.Review.
Moule RN, Jhavar SG, Eeles RA. Genotype phenotype correlation in Li-Fraumeni syndrome kindreds and its implications for management. Fam Cancer. 2006;5(2):129-33. Review.
Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles RA. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003 Oct 15;63(20):6643-50.
Strong LC. General keynote: hereditary cancer: lessons from Li-Fraumeni syndrome. Gynecol Oncol. 2003 Jan;88(1 Pt 2):S4-7; discussion S11-3. Review.
Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003 Mar;21(3):313-20. Review. Erratum in: Hum Mutat. 2003 May;21(5):551.
Vahteristo, P., Tamminen, A., Karvinen, P., Eerola, H., Eklund, C., Aaltonen, L. A., Blomqvist, C., Aittomaki, K., Nevanlinna, H. p53, CHK2, and CHK1 genes in Finnish families with Li-Fraumeni syndrome: further evidence of CHK2 in inherited cancer predisposition. Cancer Res. 61: 5718-5722, 2001.
Wong P, Verselis SJ, Garber JE, Schneider K, DiGianni L, Stockwell DH, Li FP, Syngal S. Prevalence of early onset colorectal cancer in 397 patients with classic Li-Fraumeni syndrome. Gastroenterology. 2006 Jan;130(1):73-9.