Prof.Dr. Haydar BAĞIŞ
ADYÜ Tıp Fak. Dekan Yrd.
Tıbbi Genetik ABD Başkanı
02.06.2023
Kol-Bacak Kas Distrofisi Nedenleri
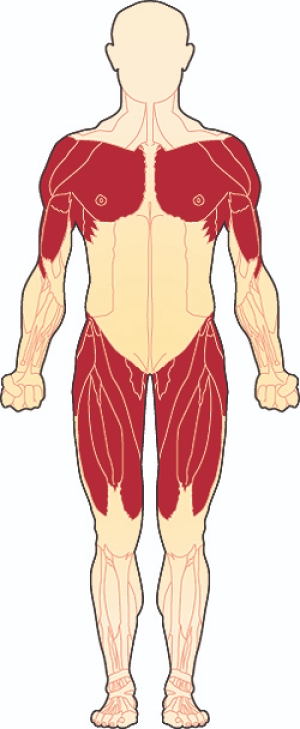
Limb-girdle musküler distrofi, kol ve bacaklardaki kasların zayıflamasına neden olan bir grup hastalık için kullanılan bir terimdir. En çok etkilenen kaslar vücuda en yakın olanlardır (proksimal kaslar), özellikle omuz, üst kol, pelvik bölge ve uyluk kaslarıdır.
Kol-bacak kas distrofisinin şiddeti, başlangıç yaşı ve özellikleri, bu durumun birçok alt tipi arasında değişiklik gösterir ve aynı aile içinde bile tutarsız olabilir. Belirti ve semptomlar ilk olarak herhangi bir yaşta ortaya çıkabilir ve bazı durumlarda hafif kalsa da genellikle zamanla kötüleşir.
Kol-bacak kas distrofisinin erken evrelerinde, etkilenen bireyler, paytak paytak veya ayak parmaklarının üzerinde yürümek gibi alışılmadık bir yürüme yürüyüşüne sahip olabilir ve ayrıca koşmada zorluk yaşayabilir. Zayıf uyluk kasları nedeniyle çömelme pozisyonundan kendilerini yukarı doğru bastırmak için kollarını kullanmaları gerekebilir. Durum ilerledikçe, uzuv kuşak kas distrofisi olan kişiler sonunda tekerlekli sandalye yardımına ihtiyaç duyabilir.
Kas erimesi, duruşta veya omuz, sırt ve kolun görünümünde değişikliklere neden olabilir. Özellikle zayıf omuz kasları, kürek kemiğinin (skapula) arkadan "dışarı çıkmasına" neden olur, bu skapular kanatlanma olarak bilinen bir işarettir. Etkilenen bireyler ayrıca anormal derecede kavisli bir bele ( lordoz ) veya yana doğru kıvrılan bir omurgaya ( skolyoz ) sahip olabilir. Bazıları kalçalarında, dizlerinde, ayak bileklerinde veya dirseklerinde hareketi kısıtlayabilen eklem sertliği (kontraktürler) geliştirir. Baldır kaslarının aşırı büyümesi (hipertrofisi), kol-kemer kas distrofisi olan bazı kişilerde görülür.
Kalp kasının zayıflaması (kardiyomiyopati), ekstremite kuşağı kas distrofisinin bazı formlarında meydana gelir. Etkilenen bazı kişiler, nefes almak için gerekli kasların zayıflığına bağlı olarak hafif ila şiddetli solunum problemleri yaşarlar. Bazı durumlarda, solunum problemleri, etkilenen bireylerin nefes almalarına yardımcı olmak için bir makine kullanmalarını gerektirecek kadar şiddetlidir (mekanik ventilasyon).
Kol-bacak kas distrofisinde zeka genellikle etkilenmez; ancak, bozukluğun nadir formlarında gelişimsel gecikme ve zihinsel yetersizlik bildirilmiştir.
SIKLIK
Kol-bacak kas distrofisinin prevalansını belirlemek zordur çünkü özellikleri değişkendir ve diğer kas bozukluklarınınkilerle örtüşür. Sıklık tahminleri 14.500'de 1 ile 123.000 kişide 1 arasında değişmektedir.
NEDENLER
Kol-bacak kas distrofisinin çeşitli formları, birçok farklı gendeki mutasyonlardan kaynaklanır. Bu genler, kas bakımı ve onarımında yer alan proteinleri yapmak için kodlama sağlar.
Bu genlerden üretilen proteinlerin bazıları, diğer proteinlerle birlikte daha büyük protein kompleksleri halinde birleşir. Bu kompleksler kas dokusunun fiziksel bütünlüğünü korur ve kasların kasılmasını sağlar. Diğer proteinler, hücre sinyalleşmesine, hücre zarı onarımına veya potansiyel olarak toksik atıkların kas hücrelerinden uzaklaştırılmasına katılır.
Kol-bacak kas distrofisi, kalıtım düzenine ve genetik nedene göre sınıflandırılır. Kol-bacakı müsküler distrofi tip 1, otozomal dominant olarak adlandırılan bir kalıtım modeline sahip bozukluğun formlarını içerir. Ekstremite-kemer kas distrofisi tip 2, otozomal resesif olarak adlandırılan bir kalıtım modeline sahip bozukluğun formlarını içerir .
Kalpainopati, proksimal ekstremite kuşak kaslarının simetrik ve ilerleyici zayıflığı ile karakterizedir. Kalpainopatinin klinik bulguları arasında parmak uçlarında yürüme eğilimi, koşmada zorluk, kürek kemiğinde kanatlanma, paytak paytak yürüme, karın kaslarında gevşeklik, Aşil tendon kısalması ve skolyoz yer alır. Etkilenen bireylerde tipik olarak kalp tutulumu veya zihinsel engel yoktur.
Kalpainopati veya ekstremite kuşağı kas distrofisi tip 2A, CAPN3 genindeki mutasyonlardan kaynaklanır . Tip 2A, ekstremite kuşağı kas distrofisinin en yaygın şeklidir ve vakaların yaklaşık yüzde 30'unu oluşturur. Kol-bacak kas distrofisi tip 2B olarak da adlandırılan disferlinopati, DYSF genindeki mutasyonlardan kaynaklanır .
Sarkoglikanopatiler, SGCA , SGCB , SGCG ve SGCD genlerindeki mutasyonların neden olduğu uzuv kuşağı müsküler distrofi formlarıdır . Bu sarkoglikanopatiler, sırasıyla 2D, 2E, 2C ve 2F tipi uzuv kuşağı kas distrofisi olarak bilinir.
Bir TTN gen mutasyonu, yalnızca Finlandiya popülasyonunda tanımlanmış olan uzuv-kuşak kas distrofisi tip 2J'ye neden olur. ANO5 genindeki mutasyonlar, uzuv kuşağı kas distrofisi tip 2L'ye neden olur. Diğer birkaç gendeki mutasyonlar, uzuv-kemer musküler distrofi tipleri 2I, 2K, 2M ve 2N dahil olmak üzere, distroglikanopatiler olarak adlandırılan uzuv-kemer kas distrofisi formlarına neden olur.
Ekstremite kuşağı kas distrofisinin diğer nadir biçimleri, bazıları tanımlanmamış olan diğer bazı genlerdeki mutasyonlardan kaynaklanır. Ek olarak, bazı araştırmacılar tarafından kol ve bacak kas distrofisi olarak sınıflandırılan belirli formlar için, diğer araştırmacılar bunları miyofibriler miyopati , Emery-Dreifuss kas distrofisi , dalgalanma kas hastalığı veya Pompe hastalığı gibi farklı, ilgili bozukluklarla gruplandırmayı önermektedir .
KALITIMI
Kol-bacak kas distrofisi farklı kalıtım modellerine sahip olabilir.
Bu durumun çoğu formu , otozomal resesif bir modelde kalıtılır ; bu, her hücredeki genin her iki kopyasının da mutasyona sahip olduğu anlamına gelir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri, mutasyona uğramış genin bir kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını göstermezler.
Bu müsküler distrofinin birkaç nadir formu, otozomal dominant bir modelde kalıtılır; bu, her hücrede değiştirilmiş genin bir kopyasının bozukluğa neden olmak için yeterli olduğu anlamına gelir.
TEDAVİ
Kol-bacak kas distrofisinde kök hücre ve gen tedavisi ile ilgili çalışmalar bulunmaktadır.
KAYNAKLAR
Understanding Neuromuscular Disease Care. IQVIA Institute. Parsippany, NJ. (2018).
Narayanaswami, P. et al. Evidence-based guideline summary?: Diagnosis and treatment of limb-girdle and distal dystrophies. Neurology (2014).
Wicklund, M. P. Limb-Girdle Muscular Dystrophies. in Encyclopedia of the Neurological Sciences (2014). doi:10.1016/B978-0-12-385157-4.00623-0
Menezes, M. P. et al. Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology (2012). doi:10.1212/WNL.0b013e318250d839
Cagliani, R. et al. A CAV3 microdeletion differentially affects skeletal muscle and myocardium. Neurology (2003). doi:10.1212/01.WNL.0000097320.35982.03
Kirschner, J. & Bönnemann, C. G. The Congenital and Limb-Girdle Muscular Dystrophies: Sharpening the Focus, Blurring the Boundaries. Archives of Neurology (2004). doi:10.1001/archneur.61.2.189
Minetti, C. et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. (1998). doi:10.1038/ng0498-365
Hauser, M. A. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum. Mol. Genet. (2002). doi:10.1093/hmg/9.14.2141
Angelini C, Giaretta L, Marozzo R. An update on diagnostic options and considerations in limb-girdle dystrophies. Expert Rev Neurother. 2018 Sep;18(9):693-703. doi: 10.1080/14737175.2018.1508997. Epub 2018 Aug 21. Citation on PubMed
Chu ML, Moran E. The Limb-Girdle Muscular Dystrophies: Is Treatment on the Horizon? Neurotherapeutics. 2018 Oct;15(4):849-862. doi: 10.1007/s13311-018-0648-x. Citation on PubMed or Free article on PubMed Central
Khadilkar SV, Patel BA, Lalkaka JA. Making sense of the clinical spectrum of limb girdle muscular dystrophies. Pract Neurol. 2018 Jun;18(3):201-210. doi: 10.1136/practneurol-2017-001799. Epub 2018 Feb 22. Citation on PubMed
Liewluck T, Milone M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018 Aug;58(2):167-177. doi: 10.1002/mus.26077. Epub 2018 Feb 7. Citation on PubMed
Liu W, Pajusalu S, Lake NJ, Zhou G, Ioannidis N, Mittal P, Johnson NE, Weihl CC, Williams BA, Albrecht DE, Rufibach LE, Lek M. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet Med. 2019 Nov;21(11):2512-2520. doi: 10.1038/s41436-019-0544-8. Epub 2019 May 20. Citation on PubMed
Mah JK, Korngut L, Fiest KM, Dykeman J, Day LJ, Pringsheim T, Jette N. A Systematic Review and Meta-analysis on the Epidemiology of the Muscular Dystrophies. Can J Neurol Sci. 2016 Jan;43(1):163-77. doi: 10.1017/cjn.2015.311. Citation on PubMed
Mitsuhashi S, Kang PB. Update on the genetics of limb girdle muscular dystrophy. Semin Pediatr Neurol. 2012 Dec;19(4):211-8. doi: 10.1016/j.spen.2012.09.008. Citation on PubMed
Straub V, Murphy A, Udd B; LGMD workshop study group. 229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017. Neuromuscul Disord. 2018 Aug;28(8):702-710. doi: 10.1016/j.nmd.2018.05.007. Epub 2018 May 24. No abstract available. Citation on PubMed
Thompson R, Straub V. Limb-girdle muscular dystrophies - international collaborations for translational research. Nat Rev Neurol. 2016 May;12(5):294-309. doi: 10.1038/nrneurol.2016.35. Epub 2016 Apr 1. Citation on PubMed
Wicklund MP, Kissel JT. The limb-girdle muscular dystrophies. Neurol Clin. 2014 Aug;32(3):729-49, ix. doi: 10.1016/j.ncl.2014.04.005. Citation on PubMed.
https://medlineplus.gov/genetics/condition/limb-girdle-muscular-dystrophy/#referenc
https://www.mda.org/disease/limb-girdle-muscular-dystrophy