Prof.Dr. Haydar BAĞIŞ
ADYÜ Tıp Fak. Dekan Yrd.
Tıbbi Genetik ABD Başkanı
10.06.2023
Hastalığa Genel Bakış
Özet
Akondroplazi, yaklaşık 20.000-30.000 canlı doğumda 1 meydana gelen, en sık görülen kemik büyümesi anormalliğidir (iskelet displazisi). Bu genetik bozukluğa, fibroblast büyüme faktörü reseptörü 3'teki ( FGFR3) bir mutasyon neden olur. Akondroplazi, hastaların yaklaşık yüzde 80'inde spontan bir genetik mutasyonun sonucu olarak ortaya çıkar; kalan yüzde 20'de bir ebeveynden miras alınır. Bu genetik bozukluk, alışılmadık derecede büyük bir kafa (makrosefali), kısa üst kollar (rizomelik cücelik) ve kısa boy (yetişkin boyu yaklaşık 4 fit) ile karakterize edilir. Akondroplazi tipik olarak zihinsel yeteneklerde bozulmaya veya eksikliklere neden olmaz. Baş ve boynu birleştiren kemikler beyin sapına veya üst omuriliğe baskı yapmazsa (kranioservikal bileşke kompresyonu), ortalama yaşam süresi normale yakındır.
Genel
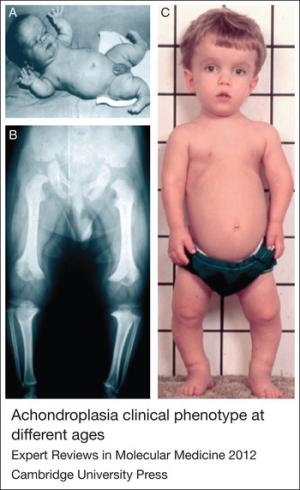
Bu nadir görülen genetik bozukluk, ayırt edici özelliklerle karakterize edilir: kısa boy (genellikle 1,2 metrenin altında); belirgin bir alın (frontal çıkıntı) ve düz (basık) burun köprüsü ile alışılmadık derecede büyük bir kafa (makrosefali); kısa kollar ve bacaklar; belirgin karın ve kalça (omurganın içe doğru kıvrımı nedeniyle); ve uzatma sırasında "trident" veya üç çatallı pozisyon alan parmakları olan kısa eller.
Bebeklik
Akondroplazi ile doğan bebeklerin tipik olarak "kubbe benzeri" (tonozlu) bir kafatası ve çok geniş bir alınları vardır. Küçük bir oranda beyin çevresinde aşırı sıvı birikimi (hidrosefali) vardır. Bebeklik dönemindeki düşük kas tonusu (hipotoni) akondroplazi için tipiktir. Gelişimsel motor kilometre taşlarının kazanılması gecikebilir.
Nedenler
Akondroplazi, fibroblast büyüme faktörü reseptörü 3 ( FGFR3 ) olarak bilinen bir genin spesifik değişikliklerinden (mutasyonlarından) kaynaklanır .
Çoğu hasta için, durumun belirgin bir aile öyküsü yoktur. Babanın artan yaşı (ileri baba yaşı), sporadik akondroplazi vakalarında katkıda bulunan bir faktör olabilir.
Akondroplazi otozomal dominant kalıtılır, yani her hücrede değiştirilmiş genin bir kopyası bozukluğa neden olmak için yeterlidir. Akondroplazili kişilerin yaklaşık yüzde 80'i, durumu olmayan ve ortalama boyda olan ebeveynlerden doğar; bu vakalara FGFR3 genindeki yeni varyantlar (de novo) neden olur . Geri kalan vakalarda, akondroplazili kişiler, etkilenen bir veya iki ebeveynden değiştirilmiş bir FGFR3 genini miras alır .
Bazı durumlarda, hipokondroplazi, belirgin bir aile öyküsü olmaksızın bilinmeyen nedenlerle (ara sıra) rastgele ortaya çıkıyor gibi görünmektedir. Diğer durumlarda, bozukluk otozomal dominant kalıtımla aileseldir. Yukarıda belirtildiği gibi hipokondroplazi ve akondroplazi aynı genin farklı mutasyonlarından kaynaklanabilir (örn.FGFR3 ).
FGFR3 geninin iki değiştirilmiş kopyasını miras alan bireyler, tipik olarak, kemiklerin aşırı derecede kısalmasına ve az gelişmiş bir göğüs kafesine neden olan ciddi bir akondroplazi formuna sahiptir. Bu kişiler genellikle ölü doğarlar veya doğumdan kısa bir süre sonra solunum yetmezliğinden ölürler.
Etkilenen popülasyonlar
Akondroplazi erkekleri ve kadınları eşit sayıda etkiliyor gibi görünmektedir. Bu bozukluk gelişmekte olan fetüste başlar ve cüceliğe neden olan en yaygın iskelet displazisi biçimlerinden biridir. Akondroplazinin tahmini sıklığı yaklaşık 15.000 doğumda bir ile 35.000 doğumda bir arasında değişmektedir.
Benzer Belirtileri Olan Bozukluklar
Aşağıdaki bozuklukların belirtileri Akondroplazininkilere benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir:
Hipokrondroplazi, küçük boy ve orantısız şekilde kısa kollar, bacaklar, eller ve ayaklar (yani kısa uzuvlu cücelik) ile karakterize edilen genetik bir bozukluktur. Bozukluğu olanlarda, boy kısalığı genellikle erken çocukluk ortalarına kadar veya bazı durumlarda yetişkinlik kadar geç fark edilmez.
Etkilenen kişilerde ayrıca erken çocukluk döneminde, genellikle yaşla birlikte kendiliğinden düzelen bacaklarda eğrilik gelişebilir. Bazı durumlarda, alışılmadık derecede büyük bir kafa (makrosefali), nispeten belirgin bir alın, dirseklerin sınırlı ekstansiyonu ve dönüşü ve/veya diğer fiziksel bulgular gibi ek anormallikler mevcut olabilir. Ek olarak, vakaların yaklaşık yüzde 10'unda hafif zeka geriliği olabilir.
Ek bozukluklar, küçük boy ve orantısız olarak kısa kollar ve bacaklar (kısa uzuvlu cücelik), başın anormal büyümesi (makrosefali), kafatası ve yüz (kraniyofasiyal) bölgesinde ek malformasyonlar ve/veya diğer benzer semptomlar ve bulgular ile karakterize edilebilir. potansiyel olarak akondroplazi ile ilişkili olanlara.
Akondroplazi, kapsamlı klinik muayene, röntgen çalışmaları ve/veya ek teşhis teknikleriyle diğer kısa uzuvlu cücelik biçimlerinden ayırt edilebilir.
Teşhis
Akondroplazinin klinik ve radyolojik özellikleri iyi karakterize edilmiştir. Tipik bulgulara sahip olanlar genellikle tanıyı doğrulamak için moleküler genetik testlere ihtiyaç duymazlar. Yenidoğanda klinik özellikler şüphe uyandırdığında, tanıyı doğrulamak için röntgen (radyografi) bulguları kullanılabilir.
Belirtiler
Bununla birlikte, belirsizlik varsa, FGFR3 geninin genetik varyantının moleküler genetik test ile tanımlanması tanıyı koymak için kullanılabilir. Aşağıda, akondroplazi tanısında kullanılabilecek klinik belirtiler:
Orantısız kısa boy
Ön çıkıntılı makrosefali
Orta yüzün geriye doğru yer değiştirmesi ve çökük burun köprüsü
Uzuvlarda fazladan deri kıvrımları olan kolların kısalması
Dirsek uzantısının sınırlandırılması
Kısaltılmış parmaklar ve ayak parmakları (brakidaktili)
Ellerin üç çatallı konfigürasyonu
Çarpık bacaklar
Omurganın abartılı içe doğru eğimi (lomber lordoz)
Eklem gevşekliği
Hidrosefali : Kafa içi basınç artışı belirtileri/semptomları ortaya çıkarsa (hızlı kafa büyümesi, şişkin fontanel, görme değişiklikleri, baş ağrısı), bir beyin cerrahına sevk gereklidir. Hidrosefali varlığını belirlemek için bebeklik döneminde beynin bilgisayarlı tomografisi (BT) veya manyetik rezonans görüntülemesi (MRG) yapılabilir.
Kranioservikal bileşke daralması: Suboksipital dekompresyon ihtiyacının öngörücüleri, bir tıp uzmanı tarafından değerlendirme gerektirir. Semptomatik kompresyon endikasyonu, bir beyin cerrahına acil sevk gerektirir.
Obstrüktif uyku apnesi : Kilo verme, bademcikleri ve adenoidleri çıkarmak için ameliyat (adenotonsillektomi), pozitif hava yolu basıncı ve nadiren boyunda bir açıklık oluşturmak için ameliyat (trakeostomi) ile tedavi edilebilir.
Orta kulak disfonksiyonu: Sık orta kulak enfeksiyonlarını yönetmek ve potansiyel işitme kaybını önlemek için yedi veya sekiz yaşına kadar kulak tüplerine ihtiyaç duyulabilir.
Kısa boy : Büyüme hormonu kullanımı üzerine yapılan araştırmalar, başlangıçta büyümenin hızlandığını, ancak zamanla azalan etki ve çok az kalıcı fayda olduğunu göstermiştir.
Obezite : Obeziteyi önlemek için önlemler erken çocukluk döneminde başlamalıdır. İlerlemeyi izlemek için akondroplaziye özgü standart ağırlık-boy ızgaraları kullanılmalıdır.
Varus deformitesi: Bacaklarda semptomatik eğrilik (varus deformitesi) bir ortopediste sevk gerektirir. Bununla birlikte, asemptomatik eğilme genellikle cerrahi düzeltmeyi garanti etmez.
Omurga şekil bozuklukları: Yaşamın ilk 12-18 ayında desteksiz oturmanın yasaklanması gibi önleyici tedbirler, omurganın ortasında sabit bir geriye eğri (kifoz) gelişme riskini azaltır. Önleyici tedbirler başarısız olursa, böyle bir deformitenin ciddiyet derecesine bağlı olarak korse veya ameliyat gerekli olabilir.
Spinal stenoz: Spinal stenoz belirtileri/belirtileri ortaya çıkarsa, acil cerrahi sevk uygundur.
Bağışıklama: Tüm rutin bağışıklamalar gereklidir.
Psikolojik Destek : Kısa boy için uyum sağlamak için ev ve okuldaki çevresel değişiklikler gerekli olabilir.
Sosyalleşme: Akondroplazili hastalar sosyalleşme ve okula uyumda zorluklarla karşılaşabilirler. Destek grupları (Little People of America gibi), akran desteği, kişisel örnek olma ve sosyal farkındalık programları yoluyla ailelere bu konularda yardımcı olabilir.
2021'de Voxzogo (vosoritide), büyüme potansiyeline izin veren akondroplazi ve açık epifizleri (büyüme plakaları) olan beş yaş ve üstü çocuklar için onaylandı.
KAYNAKLAR
TEXTBOOKS
Pauli RM, Botto LD (2018) Achondroplasia. In: Management of Genetic Syndromes. 4 ed. New York, NY: John Wiley & Sons. In press.
JOURNAL ARTICLES
Miccoli M, Bertelloni S, Massart F. Height outcome of recombinant human growth hormone treatment in achondroplasia children: a meta-analysis. Horm Res Paediatr. 2016;86:27–34.
INTERNET
Pauli RM, Legare JM. Achondroplasia. 1998 Oct 12 [Updated 2018 May 10]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018.
Bober MB, Bellus GA, Nikkel SM, et al. Hypochondroplasia. 1999 Jul 15 [Updated 2013 Sep 26]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington,
https://www.cambridge.org/core/journals/expert-reviews-in-molecular-medicine/article/abs/fgfr3-targeting-strategies-for-achondroplasia/4DE918F2A18501449E543DD599B60E14.
Horton WA, Salon JG, Hecht JT. Akondroplazi. Lancet. 2007 Temmuz 14;370(9582):162-172. doi: 10.1016/S0140-6736(07)61090-3