Prof.Dr.Haydar BAĞIŞ
ADYÜ Tıp Fak.Dekan Yrd.
Tıp Fak.Tıbbi Genetik ABD. Bşk.
15.08.2023
Özet
-
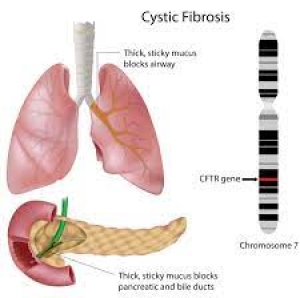
Kistik fibroz, kistik fibroz transmembran iletkenlik düzenleyici (CFTR) proteinini üreten gendeki mutasyonlardan kaynaklanır.
-
KF'li kişilerde, CFTR genindeki mutasyonlar, akciğer hücrelerinde ve vücudun diğer kısımlarında bulunan CFTR proteininin normal üretimini veya işleyişini bozabilir.
-
Kistik fibroz, resesif bir hastalığa örnektir. Bu, bir kişinin CF'ye sahip olması için CFTR geninin her iki kopyasında da bir mutasyona sahip olması gerektiği anlamına gelir.
Kistik fibroz, vücudun birçok organına zarar verebilecek kalın, yapışkan mukus birikmesi ile karakterize edilen kalıtsal bir hastalıktır. Bozukluğun en yaygın belirti ve semptomları, solunum sisteminde ilerleyici hasar ve kronik sindirim sistemi problemlerini içerir. Bozukluğun özellikleri ve şiddeti, etkilenen bireyler arasında değişir.
Mukus, hava yollarının, sindirim sisteminin, üreme sisteminin ve diğer organ ve dokuların mukozolarını kayganlaştıran ve koruyan kaygan bir maddedir. Kistik fibrozlu hastalarda vücut, anormal derecede kalın ve yapışkan mukus üretir. Bu anormal mukus , solunum yollarını tıkayarak ciddi solunum sorunlarına ve akciğerlerde bakteriyel enfeksiyonlara yol açabilir. Bu enfeksiyonlar kronik öksürüğe, hırıltıya ve iltihaba neden olur. Zamanla, mukus birikmesi ve enfeksiyonlar, akciğerlerde skar dokusu (fibroz) ve kist oluşumu dahil olmak üzere kalıcı akciğer hasarına neden olur.
Kistik fibrozlu çoğu insanda sindirim sorunları da vardır. Etkilenen bazı bebeklerde, doğumdan kısa bir süre sonra ortaya çıkan bir bağırsak tıkanıklığı olan mekonyum ileusu vardır. Diğer sindirim sorunları, pankreasta kalın, yapışkan mukus birikmesinden kaynaklanır. Pankreas, insülin (kan şekeri düzeylerini kontrol etmeye yardımcı olan bir hormon) üreten bir organdır. Ayrıca yiyecekleri sindirmeye yardımcı olan enzimler üretir. Kistik fibrozisli kişilerde mukus sıklıkla pankreasa zarar vererek onun insülin ve sindirim enzimleri üretme yeteneğini bozar. Sindirim sorunları ishale, yetersiz beslenmeye, zayıf büyümeye ve kilo kaybına yol açabilir. Ergenlikte veya yetişkinlikte, insülin eksikliği, kistik fibroz ile ilişkili diabetes mellitus olarak bilinen bir diyabet türüne neden olabilir.
Kistik fibrozis eskiden ölümcül bir çocukluk hastalığı olarak kabul edilirdi. İyileştirilmiş tedaviler ve hastalığı yönetmenin daha iyi yolları ile, kistik fibrozlu birçok kişi artık yetişkinliğe kadar iyi yaşıyor. Kistik fibrozlu yetişkinler, solunum, sindirim ve üreme sistemlerini etkileyen sağlık sorunları yaşarlar. Kistik fibrozlu çoğu erkekte , sperm taşıyan tüplerin (vaz deferens) mukus tarafından bloke edildiği ve düzgün gelişmediği bir durum olan doğuştan iki taraflı vas deferens yokluğu (CBAVD) vardır. CBAVD'li erkekler tedavi görmedikçe çocuk sahibi olamazlar (kısır). Kistik fibrozlu kadınlar gebelikte komplikasyonlar yaşayabilir.
Doğuştan iki taraflı vas deferens yokluğu (CBAVD) olan erkeklerde yaklaşık 80 CFTR mutasyonu tanımlanmıştır. Etkilenen erkeklerin çoğu, her hücrede genin en az bir kopyasında hafif bir mutasyona sahiptir. Bu mutasyonlar, CFTR proteininin işlevinin bir kısmını korumasına izin verir. Etkilenen bazı erkeklerde, her hücrede CFTR geninin bir kopyasında hafif bir mutasyon ve genin diğer kopyasında daha şiddetli, kistik fibroza neden olan bir mutasyon vardır.
CFTR genindeki mutasyonlar, klorür kanalının işlevini bozarak, klorür iyonlarının ve suyun hücrelere girip çıkmasını engeller. Sonuç olarak, erkek genital sistemindeki hücreler anormal derecede kalın ve yapışkan mukus üretir. Bu mukus, testislerden (vaz deferens) sperm taşıyan tüpleri oluşurken tıkar ve doğumdan önce bozulmalarına neden olur. Vas deferens olmadan sperm testislerden meninin bir parçası olmak üzere taşınamaz. Doğuştan iki taraflı vas deferens yokluğu olan erkekler, yardımcı üreme teknolojilerini kullanmadıkça çocuk sahibi olamazlar (kısır). Ülkemizde en yaygın mutasyon, tüm mutasyonların yaklaşık %70'ini oluşturan delta F508'dir.
Kistik fibroz, Amerika Birleşik Devletleri'ndeki beyaz popülasyonda yaygın bir genetik hastalıktır. Hastalık 2.500 ila 3.500 beyaz yenidoğanda 1'de görülür. Kistik fibroz, diğer etnik gruplarda daha az yaygındır ve yaklaşık 17.000 Afrikalı Amerikalıdan 1'ini ve 31.000 Asyalı Amerikalıdan 1'ini etkiler.
CFTR (CF transmembrane conductance regulator) genindeki mutasyonlar kistik fibroza neden olur. CFTR geni , klorür iyonları adı verilen negatif yüklü parçacıkları hücrelerin içine ve dışına taşıyan bir kanal yapmak için kodlama sağlar. Klorür, terde bulunan yaygın bir tuz olan sodyum klorürün bir bileşenidir. Klorürün hücrelerde de önemli görevleri vardır; örneğin, klorür iyonlarının akışı, ince, serbestçe akan mukus üretimi için gerekli olan suyun dokulardaki hareketini kontrol etmeye yardımcı olur.
CFTR genindeki mutasyonlar, klorür kanallarının işlevini bozarak, klorür iyonlarının ve suyun hücre zarları boyunca akışını düzenlemelerini engeller. Sonuç olarak, akciğerlerin, pankreasın ve diğer organların geçiş yollarını çevreleyen hücreler, alışılmadık derecede kalın ve yapışkan olan mukus üretir. Bu mukus, hava yollarını ve çeşitli kanalları tıkayarak kistik fibrozun karakteristik belirti ve semptomlarına neden olur.
Diğer genetik ve çevresel faktörler muhtemelen durumun ciddiyetini etkiler. Örneğin, CFTR dışındaki genlerdeki mutasyonlar, kistik fibrozlu bazı kişilerin neden diğerlerinden daha ciddi şekilde etkilendiğini açıklamaya yardımcı olabilir. Bununla birlikte, bu genetik değişikliklerin çoğu tanımlanmamıştır.
Kistik fibrosiz hastalığı , otozomal resesif bir modelde kalıtılır, yani her hücrede genin her iki kopyası da mutasyona sahiptir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri, mutasyona uğramış genin bir kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını göstermezler.
Kistik Fibroz için yeni çığır açan tedavi onayladı:
ABD Gıda ve İlaç İdaresi (FDA) bugün, en yaygın kistik fibroz mutasyonuna sahip hastaları tedavi etmek için mevcut olan ilk üçlü kombinasyon tedavisi olan Trikafta'yı (elexacaftor/ivacaftor/tezacaftor) onayladı. Trikafta, kistik fibroz popülasyonunun %90'ını temsil ettiği tahmin edilen kistik fibroz transmembran iletkenlik düzenleyici (CFTR) geninde en az bir F508del mutasyonu olan kistik fibrozlu 12 yaş ve üstü hastalar için onaylanmıştır. CFTR geninin bilinen yaklaşık 2.000 mutasyonu bulunurken, en yaygın mutasyon F508del mutasyonudur.
Trikafta, kusurlu CFTR proteinini hedefleyen üç ilacın bir kombinasyonudur. CFTR gen mutasyonu tarafından yapılan proteinin daha etkin çalışmasına yardımcı olur. Şu anda kusurlu proteini hedef alan mevcut tedaviler, kistik fibrozlu bazı hastalar için tedavi seçenekleridir, ancak birçok hasta tedavi için uygun olmayan mutasyonlara sahiptir. Trikafta, kistik fibrozlu popülasyonun %90'ını veya Amerika Birleşik Devletleri'nde yaklaşık 27.000 kişiyi etkileyen, en az bir F508del mutasyonu olan 12 yaş ve üstü kistik fibroz hastaları için etkili olan ilk onaylanmış tedavidir.
Kistik fibroza sahip olmak için, bir çocuğun her ebeveynden kistik fibroz transmembran iletkenlik düzenleyici (CFTR) gen mutasyonunun bir kopyasını alması gerekir. Bir CFTR gen mutasyonunun yalnızca bir kopyasına sahip kişilerde KF yoktur. Bunlara "CF taşıyıcıları" denir.
İki KF taşıyıcısının her çocuğu doğduğunda, şans şu şekildedir:
Çocuğun yüzde 25'i (4'te 1) CF'ye sahip olacak
Yüzde 50 (2'de 1) çocuk taşıyıcı olacak ama KF'si olmayacak
Yüzde 25 (4'te 1) çocuk genin taşıyıcısı olmayacak ve CF'ye sahip olmayacak
KF'li kişiler, CF gen mutasyonlarının kopyalarını da çocuklarına aktarabilirler. CF'li bir kişinin CF taşıyıcısı olan bir çocuğu varsa, olasılıklar:
Yüzde 50 (2'de 1) çocuk taşıyıcı olacak ama KF'si olmayacak
Yüzde 50 (2'de 1) çocuk KF'ye sahip olacak.
Bebeğim KF Taşıyıcı İse Bu Ne anlama Gelir?
Bebeğinizin yenidoğan tarama sonuçları bir KF gen mutasyonu bulduysa ve ter testi negatifse (normal), bebeğinizde KF yoktur. Bir mutasyonun test sonucu, bebeğinizin bir KF taşıyıcısı olduğu anlamına gelir. KF taşıyıcısı sağlıklıdır ve hastalığı yoktur.
Genetiği anlayacak yaşa geldiğinde çocuğunuzla konuşmanızı öneririz. Çocuğunuzun KF gen mutasyonunu gelecekteki çocuklarına geçirebileceğini bilmesi önemli olacaktır. Eşi, çocuk sahibi olmayı planlıyorsa, KF taşıyıcı testi yaptırmak isteyebilir.
KF gen mutasyonları ailelerde paylaşılır. Bu nedenle, kan bağı olan akrabalarınıza KF gen mutasyonunun taşıyıcıları olabileceklerini söylemelisiniz, böylece onlar ve eşleri test edilmek isteyip istemeyeceklerine karar verebilirler. Bu durumda tıbbi genetik uzmanından danışmanliık alınır.
Aileniz bu konuşmaya hazır olduğunda, siz ve çocuğunuz KF taşıyıcısı olmayı çocuğunuzun doktoru, genetik danışmanı veya genetik hemşiresi uzmanıyla tartışabilirsiniz.
12 yaş ve üzerindeki kistik fibrozlu hastalarda Trikafta'nın etkinliği iki çalışmada gösterilmiştir. İlk deneme, F508del mutasyonu ve ikinci alelde mutasyona sahip 403 hastada CFTR proteini olmaması veya yanıt vermeyen bir CFTR proteini ile sonuçlanan 24 haftalık, randomize, çift kör, plasebo kontrollü bir çalışmaydı. ivacaftor veya tek başına tezacaftor/ivacaftor. İkinci deneme, iki özdeş F508del mutasyonuna sahip 107 hastada dört haftalık, randomize, çift kör, aktif kontrollü bir çalışmaydı.
Her denemede, birincil analiz, kistik fibroz akciğer hastalığı ilerlemesinin yerleşik bir belirteci olan ppFEV1 olarak bilinen, bir saniye içinde tahmin edilen zorlu ekspiratuar hacim yüzdesindeki artışlara baktı. Trikafta her iki denemede de ppFEV1'i yükseltti. İlk denemede, plaseboya kıyasla ortalama ppFEV1'i başlangıca göre %13,8 artırdı. İkinci denemede, tezacaftor/ivacaftor'a kıyasla ortalama ppFEV1'i başlangıca göre %10 artırdı. İlk denemede, Trikafta ile tedavi ayrıca plaseboya kıyasla ter klorür, pulmoner alevlenmelerin sayısı (kötüleşen solunum semptomları ve akciğer fonksiyonu) ve vücut kitle indeksinde (ağırlık-boy oranı) iyileşmelerle sonuçlandı.
Trikafta'nın güvenlik profili, iki çalışmadaki 510 kistik fibroz hastasından alınan verilere dayanmaktadır. Güvenlik profili genel olarak tüm hasta alt gruplarında benzerdi. Trikafta alan hastalarda plaseboya kıyasla daha sık meydana gelen ciddi advers ilaç reaksiyonları, döküntü ve grip (grip) olaylarıdır. En yaygın advers ilaç reaksiyonları arasında baş ağrıları, üst solunum yolu enfeksiyonları, karın ağrıları, ishal, döküntüler, artmış karaciğer enzimleri (alanin aminotransferaz ve aspartat aminotransferaz), burun tıkanıklığı, artmış kan kreatin fosfokinaz (kas hasarı ile ilişkili olabilen bir enzim) yer alır. , rinore (burun boşluğunda mukus), rinit (burun mukoza zarının şişmesi), grip, sinüzit ve artmış kan bilirubini (karaciğerle ilgili problemlerden kaynaklanabilir,
Trikafta reçeteleme bilgileri, karaciğer fonksiyon testlerinin (transaminazlar ve bilirubin) yükselmesi, Sitokrom P450 3A4 (CYP3A) adı verilen başka bir karaciğer enziminin indükleyicileri veya inhibitörleri olan diğer ürünlerle birlikte kullanım ve katarakt riski ile ilgili uyarıları içerir. Hastalar ve bakıcıları, tedaviye başlamadan önce bu riskler ve aldıkları ilaçlar hakkında bir sağlık uzmanıyla konuşmalıdır.
Kistik fibrozlu hastalar bir sağlık uzmanıyla konuşmalı ve hangi gen mutasyonlarına sahip olduklarını anlamak için testler yaptırmalıdır. En az bir F508del mutasyonunun varlığı, tedaviden önce FDA onaylı bir genotipleme testi kullanılarak doğrulanmalıdır. 12 yaşından küçük kistik fibrozlu hastalarda Trikafta'nın güvenliliği ve etkinliği belirlenmemiştir.
Hastalığın Rutin İzlenmesi
Kistik fibrozlu hastalar, kısmen klinik durumlarına göre yönlendirilen ve ayrıca hastalığın standart rutin izlenmesi olarak çok sayıda incelemeye tabi tutulacaktır. Standart araştırmalar şunları içerir:
- Göğüs radyografisi: hiperinflasyonu değerlendirmek için bronş kalınlaşmasına dair kanıtlar olabilir (yıllık değerlendirmenin bir parçası olarak yıllık olarak alınır) Bazı birimler BT taraması yapar.
- Klorür ter testi (tanı sırasında ve CFTR güçlendirici/düzeltici tedavi alıyorsa yıllık)
- Mikrobiyolojik değerlendirme , örneğin öksürük sürüntüsü/balgam örneği (her klinik karşılaşmada)
- Glikoz tolerans testi (ergenlik döneminde ve sonrasında yıllık değerlendirmede)
- Karaciğer fonksiyon testi ve pıhtılaşma (yıllık değerlendirmede)
- Kemik profili (yıllık değerlendirmede)
- Akciğer fonksiyon testi – spirometri / akciğer temizleme indeksi
Hasta ve Aile Eğitimi
Hava Yolu Temizliği ve Göğüs Semptomlarının Yönetimi: Genellikle günde iki kez fizyoterapiye ihtiyaç duyulur. Fizyoterapinin mantığı, hava yolu tıkanıklığını azaltacak ve enfeksiyon riskini en aza indirecek şekilde hava yolu sekresyon klirensini arttırmaktır.
Mukolitikler ve DNaz: DNaz (dornaz alfa) inhale edilir ve KF'li hastaların balgamında bol miktarda bulunan DNA'yı sindirerek mukusun viskozitesini azaltır.
Hipertonik salin hava yolunun temizlenmesine yardımcı olabilir (ve daha fazla temizlenmesine yardımcı olmak için fizyoterapi sırasında kullanılabilir) (12 yaşın altında kullanımı destekleyen sınırlı kanıt).
Pankreas yetmezliği olan hastalarda yağ içeren öğünlerle (yemeğin başında veya yemek sırasında alınmalıdır) pankreatik enzim takviyesi (Creon) almaları gerekecektir.
Komplikasyonlar
Yine, KF'nin çoklu sistem doğası nedeniyle ortaya çıkabilecek bir dizi komplikasyon vardır.
Solunum sistemi
- Alerjik bronkopulmoner aspergilloz (ABPA) – Aspergillus spp.'nin varlığına karşı bir bağışıklık tepkisi. Spesifik tanı kriterleri vardır. ABPA başlangıçta oral kortikosteroidlerle (prednizolon) tedavi edilebilir ve itrakonzaol de denenebilir.
- Bronşektazi
- Hemoptizi oluşabilir
- Pulmoner hipertansiyon ve sağ kalp suşu
- Pnömotoraks , KF'li daha büyük çocukların %1'e kadarında görülen ve genellikle daha ilerlemiş hastalıkla ilişkilendirilen hayatı tehdit eden bir komplikasyondur. Nüks sıklıkla meydana gelir.
- KF'li hastalarda eninde sonunda solunum yetmezliği ortaya çıkacaktır.
- Nazal polipler, sinüzit ile ilişkili olabilecek KF'li çocukların %10'a kadarında görülebilir.
Gastrointestinal sistem
- Rektal prolapsus meydana gelebilir. Kesin olarak altta yatan mekanizma bilinmemektedir, ancak hacimli dışkıların sık geçişinin bir rolü olduğu öne sürülmektedir. Tedavinin ilk basamağı, çocuğun yeterli pankreatik enzim replasmanı ve ıkınmayı en aza indirmek için bir müshil almasını sağlamaktır.
- Distal intestinal obstrüksiyon sendromu (DIOS), distal ileumun tıkanmasıdır ve KF'li çocukların %10'a kadarını etkiler. DIOS'un daha yavaş bağırsak geçişine bağlı olduğu ileri sürülmektedir. Çocuk büyük olasılıkla kolik karın ağrısı ile başvuracak ve klinik muayenede RQ'da ele gelen bir kitle ortaya çıkacaktır.
- KF ile ilişkili karaciğer hastalığı
TÜM HASTALARIMIZA GEÇMİŞ OLSUN.
KAYNAKLAR:
- https://teachmepaediatrics.com/respiratory/lower-respiratory-tract/cystic-fibrosis/
- https://www.cff.org/intro-cf/cf-genetics-basics
- https://www.fda.gov/news-events/press-announcements/fda-approves-new-breakthrough-therapy-cystic-fibrosis
- https://medlineplus.gov/genetics/condition/cystic-fibrosis/#references
- Accurso FJ. Update in cystic fibrosis 2005. Am J Respir Crit Care Med. 2006 May 1;173(9):944-7. doi: 10.1164/rccm.2601006. No abstract available. Citation on PubMed or Free article on PubMed Central
- ACOG Committee Opinion No. 486: Update on carrier screening for cystic fibrosis. Obstet Gynecol. 2011 Apr;117(4):1028-1031. doi: 10.1097/AOG.0b013e31821922c2.
- Deignan JL, Astbury C, Cutting GR, Del Gaudio D, Gregg AR, Grody WW, Monaghan KG, Richards S; ACMG Laboratory Quality Assurance Committee. CFTR variant testing: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020 Aug;22(8):1288-1295. doi: 10.1038/s41436-020-0822-5. Epub 2020 May 14.
- Gardner J. What you need to know about cystic fibrosis. Nursing. 2007 Jul;37(7):52-5. doi: 10.1097/01.NURSE.0000279437.30155.1e.
- Gershman AJ, Mehta AC, Infeld M, Budev MM. Cystic fibrosis in adults: an overview for the internist. Cleve Clin J Med. 2006 Dec;73(12):1065-74. doi: 10.3949/ccjm.73.12.1065.
- Grody WW, Thompson BH, Gregg AR, Bean LH, Monaghan KG, Schneider A, Lebo RV. ACMG position statement on prenatal/preconception expanded carrier screening. Genet Med. 2013 Jun;15(6):482-3. doi: 10.1038/gim.2013.47. Epub 2013 Apr 25.
- Merlo CA, Boyle MP. Modifier genes in cystic fibrosis lung disease. J Lab Clin Med. 2003 Apr;141(4):237-41. doi: 10.1067/mlc.2003.29.
- Ratjen F, Doring G. Cystic fibrosis. Lancet. 2003 Feb 22;361(9358):681-9. doi: 10.1016/S0140-6736(03)12567-6. Citation on PubMed
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005 May 12;352(19):1992-2001. doi: 10.1056/NEJMra043184. No abstract available.
- Savant A, Lyman B, Bojanowski C, Upadia J. Cystic Fibrosis. 2001 Mar 26 [updated 2023 Mar 9]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from http://www.ncbi.nlm.nih.gov/books/NBK1250/ Citation on PubMed