
Waardenburg Sendromu Genetiği
Prof.Dr.Haydar BAĞIŞ
ADYÜ Tıp Fak.Dekan Yrd.
Tıp Fak.Tıbbi Genetik ABD. Bşk.
18.07.2023
Waardenburg sendromu (WS), adını ilk kez 1947'de işitme kaybı, distopya cantorum (yani, gözlerin iç kantisinin yana doğru yer değiştirmesi) ve retinal pigment farklılıkları olan bir hastayı tanımlayan Hollandalı göz doktoru Petrus Johannes Waardenburg'dan almıştır. 1951'de, benzer semptomları olan diğer hastaları belirledikten sonra, Waardenburg sendromu artık WS tip 1 (WS1) olarak sınıflandırmıştır.
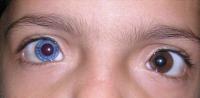
Waardenburg sendromu, işitme kaybına ve saç, cilt ve gözlerin renginde değişikliklere (pigmentasyon) neden olabilen bir grup genetik durumdur. Waardenburg sendromlu çoğu kişinin normal işitmesi olmasına rağmen, bir veya her iki kulakta orta ila ileri derecede işitme kaybı meydana gelebilir. İşitme kaybı doğuştan mevcuttur. Bu duruma sahip kişiler genellikle çok soluk mavi gözlere veya bir mavi göz ve bir kahverengi göz gibi farklı renkli gözlere sahiptir. Bazen bir gözde iki farklı renkte segment bulunur. Belirgin saç rengi (beyaz saç ya da griye dönen saç gibi) durumun başka bir yaygın belirtisidir. Waardenburg sendromunun özellikleri, etkilenen bireyler arasında, hatta aynı aileden insanlar arasında bile farklılık gösterir.
Fiziksel özellikleriyle ve bazen de genetik nedenleriyle ayırt edilen, bilinen dört Waardenburg sendromu türü vardır.
Tip I ve II çok benzer özelliklere sahiptir, ancak tip I'e sahip kişilerin neredeyse her zaman geniş aralıklı görünen gözleri vardır ve tip II'ye sahip kişilerde yoktur. Ek olarak, işitme kaybı tip II olan kişilerde tip I olanlara göre daha sık görülür.
Tip III (bazen Klein-Waardenburg sendromu olarak adlandırılır), işitme kaybı ve pigmentasyondaki değişikliklere ek olarak kol ve ellerdeki anormallikleri içerir.
Tip IV (Waardenburg-Hirschsprung hastalığı veya Waardenburg-Shah sendromu olarak da bilinir) , hem Waardenburg sendromunun hem de şiddetli kabızlığa veya bağırsak tıkanmasına neden olan bir bağırsak hastalığı olan Hirschsprung hastalığının belirti ve semptomlarına sahiptir.
Waardenburg sendromu, tahminen 40.000 kişiden 1'ini etkiler. Tüm doğuştan işitme kaybı vakalarının yüzde 2 ila 5'ini oluşturur. Tip I ve II, Waardenburg sendromunun en yaygın formlarıdır, tip III ve IV ise nadirdir.
EDN3, EDNRB, MITF, PAX3, SNAI2 ve SOX10 genlerindeki varyantlar (mutasyonlar olarak da bilinir) Waardenburg sendromuna neden olabilir. Bu genler, melanosit adı verilen pigment üreten hücreler de dahil olmak üzere çeşitli hücre türlerinin oluşumunda ve gelişiminde rol oynar.
Melanositler, cilt, saç ve göz rengine katkıda bulunan ve iç kulağın normal işlevinde önemli bir rol oynayan melanin adlı bir pigment üretir. Bu genlerin herhangi birindeki varyantlar, melanositlerin normal gelişimini bozarak ciltte, saçta ve gözlerde anormal pigmentasyona ve işitme sorunlarına yol açar.
Waardenburg sendromu tip I ve III, PAX3 genindeki varyantlardan kaynaklanır . MITF veya SNAI2 genindeki varyantlar, Waardenburg sendromu tip II'ye neden olabilir.
SOX10 , EDN3 veya EDNRB genindeki varyantlar, Waardenburg sendromu tip IV'e neden olabilir. Melanosit gelişimine ek olarak, bu genler kalın bağırsakta sinir hücrelerinin gelişimi için önemlidir. Bu genlerin herhangi birindeki varyantlar, işitme kaybına, pigmentasyonda değişikliklere ve Hirschsprung hastalığına bağlı bağırsak problemlerine neden olur.
Bazı durumlarda, Waardenburg sendromunun genetik nedeni tespit edilememiştir.
Waardenburg sendromu genellikle otozomal dominant olarak kalıtılır, yani her hücrede değiştirilmiş genin bir kopyası bozukluğa neden olmak için yeterlidir. Çoğu durumda, etkilenen bir kişinin durumu olan bir ebeveyni vardır. Vakaların küçük bir yüzdesi, gendeki yeni varyantlardan kaynaklanır ; bu vakalar, ailelerinde hastalık öyküsü olmayan kişilerde görülür.
Bazı Waardenburg sendromu tip II ve tip IV vakalarının otozomal resesif kalıtım modeline sahip olduğu görülmektedir , bu da her hücrede genin her iki kopyasının da varyantları olduğu anlamına gelir. Çoğu zaman, otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri değiştirilmiş genin bir kopyasını taşır, ancak durumun belirtilerini ve semptomlarını göstermez.
KAYNAKLAR
https://lmhofmeyr.co.za/conditions/ear-problems/waardenburg-syndrome/
https://emedicine.medscape.com/article/950277-overview
https://medlineplus.gov/genetics/condition/waardenburg-syndrome/#references
Huang S, Song J, He C, Cai X, Yuan K, Mei L, Feng Y. Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome. Gene Ther. 2022 Sep;29(9):479-497.
Nayak CS, Isaacson G. Worldwide distribution of Waardenburg syndrome. Ann Otol Rhinol Laryngol. 2003 Sep;112(9 Pt 1):817-20.
Newton VE. Clinical features of the Waardenburg syndromes. Adv Otorhinolaryngol. 2002;61:201-8.
Pardono E, van Bever Y, van den Ende J, Havrenne PC, Iughetti P, Maestrelli SR, Costa F O, Richieri-Costa A, Frota-Pessoa O, Otto PA. Waardenburg syndrome: clinical differentiation between types I and II. Am J Med Genet A. 2003 Mar 15;117A(3):223-35.
Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010 Apr;31(4):391-406.
Zaman A, Capper R, Baddoo W. Waardenburg syndrome: more common than you think! Clin Otolaryngol. 2015 Feb;40(1):44-8.
- Ailesel Hiperkolesterolemi Genetik Yaklaşım
- Kalıtsal Polikistik Böbrek Hastalığına Genetik Yaklaşım
- Friedreich Ataksisi Genetiği
- Şizofreninin Genetik Yönü Varmıdır ?????
- Uzun Ömürde Epigenetik Organ Yaşınızın Rolü
- Waardenburg Sendromu Genetiği
- Göz Renginde Genetiğin Rolü Nedir ?
- Noonan Sendromuna Genetik Yaklaşım.
- Akondroplazi (Cücelik) Genetiği
- Hipohidrotik Ektodermal Displazi Genetiği
- Kistik Fibrosiz Genetiği ve Tedavisinde En Son Gelişmeler
- Adrenolökodistrofi (X-ALD) ( Lorenzo’nun Yağı)
- Hemofili Genetiğinde Gen Tedavisi
- X'e Bağlı Öldürücü İnfantil SMA-2 (X'e Bağlı Spinal Müsküler Atrofi-2)
- Prof. Dr. Bağış: Trafik Kazası Dışındaki Her Hastalık Genetiktir
- Prof.Dr.Haydar Bağış’ın İsmi Doğum Yeri BESNİ'de Bir Caddeye Verildi.
- Li-Fraumeni Sendromu (LFS) Genetiği
- Cowden Sendromu (PTEN Hamartom Tümör Sendromu)
- Limb-Girdle Musküler Distrofi (LGMBD)( Kol-Bacak Kas Distrofisi) Genetiği
- EPİGENOM VE EPİGENETİK NEDİR
- Mesane Kanseri Genetiği
- SHOX Geni ve İdiyopatik Boy Kısalığı
- Alfa-Talasemi Mental Retardasyon Sendromu
- Albino (Albinizm) Hastalığı Genetiği
- Kromozom 10q26 delesyon sendromlu hastalar
- MTHFR Geni Hakkında Bilmeniz Gerekenler
- Ailevi Akdeniz Ateşi (FMF) Genetik Geçişli Hastalıktır
- Faktör V Leiden Mutasyonu ve Derin Ven Trombozu
- BRCA1/2 Genleri Meme Over Kanseri
- Nörofibromatosiz Tip 1 (NF1) Genetiği
- NADİR GENETİK HASTALIKLAR
- Genetik Yatkınlık Nedir ?
- D-dimer testi ve COVID-19
- Kulaklarda Franks işaretinin akut aort diseksiyon bağlantısı var mı?
- COVID-19 Aşıları Erkeklerde İktidarsızlık ve Kısırlık Yapmaz.
- MTHFR C677T, Faktör V Leiden, PAI 4G/5G, Protrombin G20210A Mutasyonu
- Geleceğin 100 Mesleği ve YKS Tercihi
- İki Doz COVID-19 Aşılı Kişilere CDC Önerileri
- COVID-19 mRNA Aşıları Hakkında Merak Etikleriniz
- COVID-19 Virüsünün Varyantları ve Aşılar
- COVID-19 Yeni Nesil Aşılar Nasıl Çalışır?
- Pıhtılaşma Bozukluğunda Factor V Leiden (FVL) Trombofili Genetiğinin Rolü
- Gençlerde Erişkin Tip Diyabette (MODY) Genetiğin Rolü
- Otizimin Genetik ve Çocukluk Aşılarıyla Bağlantısı Varmı?
- Genomik ve Genetik
- DOWN SENDROMLU HER ÇOCUĞUMUZ KESİNLİKLE KROMOZOM ANALİZİ YAPTIRMALI
- Ailesel Kanserler (Li-Fraumeni Sendromu ) Genetiği
- Doğuştan Kalp Hastalığının Genetik Yönü Varmıdır?
- KÖPEKLER COVID-19 KOKLAYARAK TESPİT ETMEYE BAŞLADI
- Tourette Sendromunda Genetiğin Rolü
- COVID-19 ile Karıştırılan Üst ve Alt Solunum Yolu Enfeksiyon Hastalıkları
- Renin – Anjiyotensin – Aldosteron Sistem Blokerleri ve Covid Riski Arasında İlişki Araştırıldı
- Omurilik Felcinde (Parapleji) Kök Hücre Yaklaşımı
- Genetik Hastalıkların Çocuklarıma Geçme Olasılığı Varmıdır?
- ADYÜ Tıbbi Genetik ve Tıbbi Genetik Uzmanlığı Eğitimi
- Geleceğin Ramboları Genom Düzenleme İle Yaratılmak mı İsteniyor?
- ÇOCUKLUK ÇAĞI AŞILARI HAKKINDA BİLENDE KONUŞUYOR BİLMEYENDE......
- Sağlığınızın Yolu Hissettiğiniz Genç Yaşa Bağlı......
- Adıyaman Üniversitesi Deney Hayvanları Sertifika Kurs Kayıtları Başlamıştır.
- En Popüler 65 Sosyal Medya Siteleri Hakkında Daha Fazla Bilgi Edinin
- 21 nci Yüzyılda Doktorları Neler Bekliyor ve Bakın Onlar Ne ile Uğraşıyorlar...........
- GENETİK DANIŞMANLIK HANGİ DURUMLARDA ALINMALI
- YÖK TARAFINDAN ARAŞTIRMA VE ADAY ARAŞTIRMA ÜNİVERSİTELERİNİN İKİ YILLIK PERFORMANSLARI AÇIKLANDI
- Nobel Fizyoloji ve Tıp 2019 Ödülünün Arka Planı
- Parkinson Hastalığının Genetik Yönü Varmıdır?
- Karaciğer Transplantasyonu Yerine Yeni Bir Kök Hücrenin Nakli.
- Adıyaman Üniversitesi (ADYÜ) Kimliği
- DOKTOR ADAYLARINI ADIYAMAN ÜNV. TIP FAK. BEKLİYORUZ.
- Dikkat eksikliği hiperaktivite bozukluğunun genetik bağlantısı?
- Erkekler Dikkat Ereksiyon Bozukluğunun Genetik Bağlantısı Ortaya Kondu.
- Şekerli içecekler ve kanser arasında olası bir bağlantı ortaya kondu
- Çölyak Hastalığının Belirtileri Nelerdir
- GENOM DÜZENLEME TEKNOLOJİSİ İLE OTİSTİK MAYMUN MODELİ
- OTİZİM ve BURUN İÇİ OKSİTOSİN UYGALAMASININ FAYDA VE ZARARLARI
- MUTLULUĞUNUZ İÇİN DOPAMİNİNİZİ NASIL ARTTIRILABİLİR.SİNİZ
- Mizacımız Genetik Kodlarımız Tarafından Belirleniyor........
- Rett Sendromu Otizm ya da Down Sendromu ile Karıştırılan bir Hastalık.
- Genetik Test Nedir?
- Eğitimde STEM Yaklaşımının Önemi.
- Bilim Adamları Y Kromozomsuz Erkek Fareler Yetiştirdiklerini Açıkladılar.
- 21 Mart Dünya Down Sendromu Günü ve Down Sendromu Nedir?
- KANSER İMMÜNOTERAPİSİ NEDİR?
- Dünyanın ilk genetik tasarımlı bebekleri Çin'de doğdu
- PARKİNSON HASTALIĞI İLE APANDİS ARASINDA İLİŞKİ BULUNDU.
- CRISPR köpeklerde Duchenne musküler distrofi ilerlemesini durdurdu.
- BAKIR ve ALZHEİMER HASTALIĞI
- Doymuş Yağ Kullanımında Bilinen Yanlışlar........
- OTİZMLE HAMİLE ANNENİN BAĞIRSAK MİKROBİYOMU ARASINDAKİ İLİŞKİ
- KANSERDE SIĞIR KIKIRDAĞI KULLANIMININ ÇOK ETİKİLİ OLDUĞU ORTAYA KONDU.
- ALKALİ SU TÜKET DAHA SAĞLIKLI YAŞA..........
- Dünyanın İlk Klon Maymunları Zhong Zhong ve Hua Hua 7 aylık oldu.
- Amerikan Ulusal Kanser ve Sağlık Enstitüleri (NIH) Hakkında Bilgi
- GENOMDAKİ TRANSPOZONLAR BENCİL PARAZİTLERMİ ?
- Sigara İçen Y kromozomunu yitiren erkeklerin, yitirmeyenlerden ortalamada 5.5 yıl daha erken öldüğü tespit edilmiş.
- FDA, Kolorektal Kanser, Lösemi ve Melanom için Yeni Tedavileri Onayladı
- BAĞIRSAK MİKROPLARININ OBEZİYETE ETKİSİ
- GENOM MÜHENDİSLİĞİNDE CRISPR/CAS9 TEKNOLOJİSİNİN DENEYSEL HAYVAN MODELLERİNDEKİ YERİ
- GEBZE TEKNİK ÜNİVERSİTESİ 2. GENETİK GÜNLERİ ETKİNLİĞİ
- 28 Şubat Nadir Hastalıklar Günü. Nadir Görülen Hastalıkların% 80'i Genetik Kökenlidir
- MEME KANSERİNİN YAYILIMINDA BİRÇOK GIDADA BULUNAN ASPARAJİNİN SORUMLU OLDUĞU TESPİT EDİLDİ.
- Kolon Kanserinde Bağırsak Bakterisinin Rolü Olduğu Anlaşıldı.
- Yeni Otizm İlaçları, Klinik Öncesi Çalışmada Umut Vaad Etti
- OTİSTİK ÇOCUKLARA GAİTA MİKROBİYOMUNUN TRANSFERİ
- ERKEKLERDE FAZLA VİTAMİN B6 VE B12 AKCİĞER KANSER RİSKİNİ ARTTIRABİLİR DİKKAT...
- CRH geninde keşfedilen epigenetik değişiklikler, intihar riskini artırıyor....
- FDA, 2. Gen Terapisini Onayladı
- Kolesterol testleri için emeklilik zamanı geldi mi?
- Her 30 Dakikada Bir Hareket Et ÖLÜM RİSKİNİ AZALT.............
- Homeobox Genleri Nelerdir......
- Genom düzenleme ve CRISPR-Cas9 nedir?
- Kalp krizi ve felç gibi şeylere karşı riskinizi öğrenmek istermisiniz.
- Cebinizdeki Çantanızdaki Akıllı Telefonunun Zararları Nelerdir?
- Tütün Yanarken Oluşan Yüzlerce Farklı Element Epigenetik Modifikasyonlara Yol Açıyor
- 2017 NOBEL TIP FİZYOLOJİ ÖDÜLÜ SİRKADİYEN RİTMİNİ KONTROL EDEN GENLERE VERİLDİ
- Diyabet İlacı Metformin Bazı Kanser Türlerine Karşı
- SMA Hastalarında Yeni ve Pahalı Tedavi Dönemi Başladı
- TÜRKİYE'de NASIL DOÇENT OLUNUR.
- Bir Günde 2 Yumurta Yemeye Başlarsanız Vücudunuza Olabilecek 9 Olumlu Şey.
- Bağırsamızdaki Mikroplar Yeni Organımız mı?
- BEBEĞİN OBEZİTE RİSKİ ANNENİN BESLENMESİNE BAĞLI OLARAK RAHİMDE BAŞLIYOR (GENLERİN AÇILIP KAPANMASINDAN BAŞLIYOR)
- Çay tüketimi kadınlarda epigenetik değişikliklere neden olduğu ortaya kondu
- İSTANBUL BESNİLİLER DAYANIŞMA DERNEĞİNDE AKRABA EVLİLİKLERİNDE GENETİK YAKLAŞIM KONFERANSI
- NATURE GENETİK DERGİSİ 25 YILDAKİ EN İYİ MAKALELERİNİ SEÇTİ
- Çölyak Hastalığında Reovirusların Rolü Ortaya Kondu
- cjCRISPR/Cas9 ile KÖRLÜK TEDAVİSİ
- CRISPR-Cas9 İLE NADİR KAN HASTALIKLARININ TEDAVİSİNDE YENİ UMUT
- NADİR GÖRÜLEN GENETİK HASTALIKLARIN TEŞHİSİNDE YÜZ TANIMA YAZILIMI
- Öğretmeni Şok Eden Cevap. Öğretmen Öğrenciye Sorar Ne Olmak İstiyorsun? Öğrenci Haydar BAĞIŞ Olmak İstiyorum Diye Cevap Verir
- UYKU YOKSUNLUĞU KALP HASTALIKLARINA DAVETİYE ÇIKARIYOR DİKKAT...........
- 8 Mart Dünya Kadınlar Gününüz Kutlu Olsun
- Etnisite Gibi Kültürel Farklılıklar, DNA Üzerinde İz Bırakabilir
- ZAMANINDA UYKU BEYNİ SIFIRLAYIP HAFIZAYI GÜÇLENDİRİP HASTALIKLARA DİRENÇLİ OLMAMIZI SAĞLIYOR.
- İnsanda Körelmiş 10 Organ
- 2016 Yılındaki En İyi 10 Tıbbi Yenilik
- FAZLA KİLOLARINIZ DNA METİLASYONUNU ETKİLER
- 10 Ocak Çalışan Gazetecilerimizin Gününü Kutlarım
- IPS KÖK HÜCRELERİ DONÖRÜN YAŞININ GENOMİK İZLERİNİ TAŞIYOR
- ÇOCUK İSTİSMARI İNTİHAR ve EPİGENETİK
- AKRABA EVLİLİĞİ GENETİK DANIŞMANLIK.........
- TIBBİ GENETİK VE KLİNİK UYGULAMALARI KİTABIMIZ RAFLARDA
- Periyodik Tablo’ya Dört Yeni Element Resmen Eklendi
- YENİ YILINIZ KUTLU VE MUTLU OLSUN
- EMBRİYONUN GENOM BÜTÜNLÜĞÜNÜN NASIL KORUNDUĞU KEŞFEDİLDİ.
- BAĞIRSAK MİKROBİYOMUNA DİKKAT YOKSA PARKİNSON OLABİLİRSİNİZ..........
- Kediler Hakkında Bilmediğimiz 10 şey
- Akciğer Kanserli Hastada İlk Defa CRISPR-Cas9 Tekniği ile Düzeltilmiş Hücre Nakli.....
- Doçentlik Başvurusu Yapan Adayların Dikkatine?
- CRISPR/Cas9 İLE SAĞLIKLI İNSAN EMBRİYO ÜRETİLMESİ
- GEBELERDE SABAH BULANTISI "'Düşük Riskini Azaltıyor'
- BAZI AĞRI KESİCİLER BAZI KİŞİLERDE KALP YETMEZLİĞİ RİSKİNİ ARTTIRABİLİR.
- SİGARA İÇENLERİN AKCİĞERİNDE 150 MUTASYON SAPTANDI.....
- Yeni bir tüp bebek yöntemiyle genetiği değiştirilmiş sağlam bir bebek dünyaya geldi
- Panorama Test - Non İnvaziv Prenatal Test (NIPT)
- GENETİKTE SIK SORULAN SORULAR
- Doğacak bebekte kalıtsal hastalık görülme riskinin öngörülmesi amacıyla yapılan genetik tarama testi nedir.
- Prof.Dr.Haydar BAĞIŞ Nadir Hastalıklar Konusunda Açıklama Yaptı.
- Genetik Uzmanı Prof. Dr. Haydar Bağış, insanların yedikleri ve düşündüklerinin genlerin çalışmasını etkilediğini kaydetti
- B Vitaminlerinin Beynin Yaşlanması Üzerine Etkileri
- ADYÜ TIP FAK. ÖĞRENCİLERİ SAĞLIK PROJESİ SUNUMLARI
- Prof.Dr.HAYDAR BAĞIŞ HABERTÜRK TV ÖTEKİ GÜNDEM PROGRAMINA CANLI YAYIN KONUĞU OLACAK
- Klonlama Teknolojisi, Türkiye İçin Yeni Bir Gelir Kapısı Olabilir
- Bilim depresyonu alt eden besteyi buldu?
- AKADEMİK TEŞVİK ÖDENEĞİ YÖNETMELİĞİ YAYINLANDI
- SIÇRAYAN GENLER BENCİL Mİ?
- Hücrenin Sayı Sayma Kabiliyeti
- EHLİYET, LİYAKAT VE ÜNİVERSİTELER……
- 2014 Yılı Ar-Ge Faaliyetleri Anketi Sonuçları
- İnsan Bedeni Hakkında Şaşırtıcı Gerçekler
- Türkiye'deki Klon Sığırlar Çoğalmaya Devam Ediyor.
- Türk Bilim Adamı Aziz Sancar Hocamız Bu yıl NOBEL Kimya Ödülüne Layık Görüldü.
- Bilim adamları Petri kabında kök hücreden minyatür insan böbreği üretmeyi başardı.
- İnsanı Felakete Götüren 10 Şey
- Klonlanmış hayvanın eti zararlı mı?
- İnsan embriyosunda genetik değişimler yapılıp, DNA'mız ile mi Oynanacak Acaba ?
- Meme Kanserine Genetik Yaklaşım
- Baştan Sona Avrupa Turunda Gezdiğimiz Ülke ve Şehirler
- KAMAG 1007 Programı Kapsamında Yükseköğretim Kurulu Başkanlığı Adına Yeni Çağrılar Açıldı
- BABALIK TESTİ (PATERNİTE) NEDİR VE NASIL YAPILIR
- TIBBİ GENETİKTE KARDİYOVASKÜLER RİSK PANELİ NEDİR?
- Prof. Dr. Haydar BAĞIŞ İnsanı Klonlamak Hayvanı Klonlamaktan Daha Kolay Diyor.
- YÖK Hastaneleri yok saydı!
- TÜBİTAK Doktoralı Kişilerin İstihdamını Teşvik Eden Yeni Bir Çağrı Açtı
- URAP: ODTÜ Enformatik Enstitüsü Yayın Değelendirme Kriterleri
- Akademisyenlerin ‘atıf’ çetesi
- Adıyaman Fen Lisesi Öğrencileri Tıp Fakültemizi ve Beni Ziyarete Geldiler
- Prof.Dr. Haydar BAĞIŞ diyor ki bebeğin cinsiyetini erkek belirliyor Toplumda sıkça kullanılarak yer edinen 'erkek adamın erkek çocuğu olur' düşüncesinin yanlış olduğu ortaya çıktı. 24.03.2011
- Prof.Dr. Haydar BAĞIŞ'ın da Bölüm yazarlığı yaptığı "Current Aplications of Biotechnology" kitabı yayınlandı.
- ÜNLÜ TİYATRO SANATÇIMIZ ZEKİ ALASYA YI KAYBETTİK
- YÖK Başkanı Prof.Dr. Yekta SARAÇ 'Üniversiteler susturulmuş' değil isteksiz
- Rektörlük Seçimleri ve Muhalefet
- Prof.Dr.Haydar BAĞIŞ'ın Görüşü YENİ YOK YASASI
- Adıyaman Üniversitesi Rektörlük Seçim Sonuçları
- REKTÖRLÜK SEÇİMİNİ BİLİMİN KAZANMASI DİLEĞİYLE
- Tüm Hocalarımıza Teşekkür Ederim
- HEDEFİMİZ; SÖZÜ DİNLENEN, DANIŞILAN, BİLGİ ÜRETEN, ULUSLARARASI SAYGINLIĞI OLAN BİR ÜNİVERSİTE OLUŞTURMAKTIR. BUNU HEP BİRLİKTE BAŞARACAĞIZ VE ÜNİVERSİTEMİZİ ORTAK AKILLA BERABER YÖNETECEĞİZ.
- Prof.Dr.Haydar BAĞIŞ REKTÖR ADAYLIĞI BAŞVURU DİLEKÇE VE FORMLARINI REKTÖRLÜĞE TESLİM ETTİ.
- Kalıtsal Hastalıklardan Temizlenmiş Tüp Bebekler
- Prof.Dr. Haydar BAĞIŞ Kitapları
- Prof. Dr. Bağış, Seçim Çalışmalarının Yanında Bilimsel Çalışmalarına Devam Ediyor
- Prof.Dr.Haydar BAĞIŞ'ın Projesi TÜBİTAK Akademik Başarı Öyküleri Kitabı’ nda Yer Aldı
- Hastalığım Süresince Arayan Tüm Dostlarıma Teşekkür Ederim.
- Tıp Eğitiminde E-Öğrenme
- Adaylara değil, partilere, liderlere oy verilecek
- Prof.Dr.Haydar BAĞIŞ SANKO Üniversitesi'nde "Moleküler Tıpta Biyomühendislik ve İnovasyon" konulu sempozyumunda konuştu..
- YDS SINAV STRATEJİSİ CÜNEYT BADEMCİ
- ÜNLÜ AKADEMİSYEN PROF. DR. HAYDAR BAĞIŞ KMÜ’LÜ ÖĞRENCİLERLE BULUŞTU
- Berber Çıraklığı Yaptım Ustamı Fatih semtinde Ziyaret Ettim.
- 14 Mart Tıp Bayramı Kutlaması.
- 8 MART DÜNYA KADINLAR GÜNÜ MESAJI
- "MOLEKÜLER TIPTA BİYOMÜHENDİSLİK VE İNOVASYON" SEMPOZYUMU
- İki kız evladı babası olarak erkek olmaktan utanç duyuyorum. !
- ADYÜ Tıp Fak. Tıbbi Genetik Anabilim Dalına Bağlı Genetik Tanı Biriminde Yılda 2500 Hastaya Genetik Test Hizmeti Verilmektedir.
- Bebeğin cinsiyetini erkek belirliyor
- Prof.Dr. Haydar BAĞIŞ ADYÜ Tıp Fak. Konferans Salonunda "Bilimsel Makale Nasıl Yazılır ve Yayınlanır" Başlıklı Seminer Verdi.
- Adıyaman Ünv. Tıp Fak. Öğrenci Sempozyumu Yaptı.
- Prof.Dr. Haydar BAĞIŞ Başkanlığındaki TÜBİTAK Ekibi Dünyada ilkez "Türk Malı Buzul Ayısı" ismini verdiği Donmaya Dirençli Transgenik Fare Soyları Geliştirildi
- Prof. Bağiş, OMÜ’de Transgenik ve Klon Hayvan Üretimini anlattı
- 8. Moleküler Biyoteknoloji Bahar Okulu, 19-22 Nisan, 2013
- Prof.Dr. Haydar BAĞIŞ Başkanlığındaki Ekip tarafından Ülkemizde İlk Defa Transgenik Zebra Balığı Yapıldı.
- Prof.Dr. Haydar BAĞIŞ'ın projesi TÜBİTAK Akademik Başarı Öyküleri Kitabı’ nda Yer aldı.
- Tıp Fakültesi Önünde Basın Bildirisi - Adıyaman 16 Eylül 2009 Çarşamba 15:03
- Prof. Dr. Haydar Bağış: Transgenik hayvan çalışmalarının bilime katkıları isimli sunum yaptı.
- Fare sütü kansere çare olacak. Donmaya dirençli, “Türk malı buzul ayısı’’ adı verilen bir fare geliştirerek adlarını duyuran TÜBİTAK araştırmacıları bir başarıya daha imza attı
- KLONLAMAYA TEPKİLER
- TÜBİTAK'tan müthiş bir proje daha TÜBİTAK, fare sütünde, özellikle kanser tedavisinde kullanılan insana ait 'interferon gamma' isimli bir protein üretti
- TÜBİTAK interferon gamma üretti
- Prof.Dr. Haydar BAĞIŞ ve Prof.Dr. Munis DÜNDAR Editör ve yazarlığını yaptığı " Modern Biyoteknoloji Uygulamaları" Başlıklı Kitap
- Konu: EŞCİNSELLİK GENETİK Mİ? PELİN ÇİFT: Prof Dr Haydar BAĞIŞ ve Prof Dr Uğur ÖZBEK-