
Limb-Girdle Musküler Distrofi (LGMBD)( Kol-Bacak Kas Distrofisi) Genetiği
Prof.Dr. Haydar BAĞIŞ
ADYÜ Tıp Fak. Dekan Yrd.
Tıbbi Genetik ABD Başkanı
02.06.2023
Kol-Bacak Kas Distrofisi Nedenleri
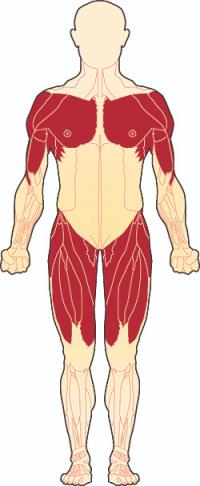
Limb-girdle musküler distrofi, kol ve bacaklardaki kasların zayıflamasına neden olan bir grup hastalık için kullanılan bir terimdir. En çok etkilenen kaslar vücuda en yakın olanlardır (proksimal kaslar), özellikle omuz, üst kol, pelvik bölge ve uyluk kaslarıdır.
Kol-bacak kas distrofisinin şiddeti, başlangıç yaşı ve özellikleri, bu durumun birçok alt tipi arasında değişiklik gösterir ve aynı aile içinde bile tutarsız olabilir. Belirti ve semptomlar ilk olarak herhangi bir yaşta ortaya çıkabilir ve bazı durumlarda hafif kalsa da genellikle zamanla kötüleşir.
Kol-bacak kas distrofisinin erken evrelerinde, etkilenen bireyler, paytak paytak veya ayak parmaklarının üzerinde yürümek gibi alışılmadık bir yürüme yürüyüşüne sahip olabilir ve ayrıca koşmada zorluk yaşayabilir. Zayıf uyluk kasları nedeniyle çömelme pozisyonundan kendilerini yukarı doğru bastırmak için kollarını kullanmaları gerekebilir. Durum ilerledikçe, uzuv kuşak kas distrofisi olan kişiler sonunda tekerlekli sandalye yardımına ihtiyaç duyabilir.
Kas erimesi, duruşta veya omuz, sırt ve kolun görünümünde değişikliklere neden olabilir. Özellikle zayıf omuz kasları, kürek kemiğinin (skapula) arkadan "dışarı çıkmasına" neden olur, bu skapular kanatlanma olarak bilinen bir işarettir. Etkilenen bireyler ayrıca anormal derecede kavisli bir bele ( lordoz ) veya yana doğru kıvrılan bir omurgaya ( skolyoz ) sahip olabilir. Bazıları kalçalarında, dizlerinde, ayak bileklerinde veya dirseklerinde hareketi kısıtlayabilen eklem sertliği (kontraktürler) geliştirir. Baldır kaslarının aşırı büyümesi (hipertrofisi), kol-kemer kas distrofisi olan bazı kişilerde görülür.
Kalp kasının zayıflaması (kardiyomiyopati), ekstremite kuşağı kas distrofisinin bazı formlarında meydana gelir. Etkilenen bazı kişiler, nefes almak için gerekli kasların zayıflığına bağlı olarak hafif ila şiddetli solunum problemleri yaşarlar. Bazı durumlarda, solunum problemleri, etkilenen bireylerin nefes almalarına yardımcı olmak için bir makine kullanmalarını gerektirecek kadar şiddetlidir (mekanik ventilasyon).
Kol-bacak kas distrofisinde zeka genellikle etkilenmez; ancak, bozukluğun nadir formlarında gelişimsel gecikme ve zihinsel yetersizlik bildirilmiştir.
SIKLIK
Kol-bacak kas distrofisinin prevalansını belirlemek zordur çünkü özellikleri değişkendir ve diğer kas bozukluklarınınkilerle örtüşür. Sıklık tahminleri 14.500'de 1 ile 123.000 kişide 1 arasında değişmektedir.
NEDENLER
Kol-bacak kas distrofisinin çeşitli formları, birçok farklı gendeki mutasyonlardan kaynaklanır. Bu genler, kas bakımı ve onarımında yer alan proteinleri yapmak için kodlama sağlar.
Bu genlerden üretilen proteinlerin bazıları, diğer proteinlerle birlikte daha büyük protein kompleksleri halinde birleşir. Bu kompleksler kas dokusunun fiziksel bütünlüğünü korur ve kasların kasılmasını sağlar. Diğer proteinler, hücre sinyalleşmesine, hücre zarı onarımına veya potansiyel olarak toksik atıkların kas hücrelerinden uzaklaştırılmasına katılır.
Kol-bacak kas distrofisi, kalıtım düzenine ve genetik nedene göre sınıflandırılır. Kol-bacakı müsküler distrofi tip 1, otozomal dominant olarak adlandırılan bir kalıtım modeline sahip bozukluğun formlarını içerir. Ekstremite-kemer kas distrofisi tip 2, otozomal resesif olarak adlandırılan bir kalıtım modeline sahip bozukluğun formlarını içerir .
Kalpainopati, proksimal ekstremite kuşak kaslarının simetrik ve ilerleyici zayıflığı ile karakterizedir. Kalpainopatinin klinik bulguları arasında parmak uçlarında yürüme eğilimi, koşmada zorluk, kürek kemiğinde kanatlanma, paytak paytak yürüme, karın kaslarında gevşeklik, Aşil tendon kısalması ve skolyoz yer alır. Etkilenen bireylerde tipik olarak kalp tutulumu veya zihinsel engel yoktur.
Kalpainopati veya ekstremite kuşağı kas distrofisi tip 2A, CAPN3 genindeki mutasyonlardan kaynaklanır . Tip 2A, ekstremite kuşağı kas distrofisinin en yaygın şeklidir ve vakaların yaklaşık yüzde 30'unu oluşturur. Kol-bacak kas distrofisi tip 2B olarak da adlandırılan disferlinopati, DYSF genindeki mutasyonlardan kaynaklanır .
Sarkoglikanopatiler, SGCA , SGCB , SGCG ve SGCD genlerindeki mutasyonların neden olduğu uzuv kuşağı müsküler distrofi formlarıdır . Bu sarkoglikanopatiler, sırasıyla 2D, 2E, 2C ve 2F tipi uzuv kuşağı kas distrofisi olarak bilinir.
Bir TTN gen mutasyonu, yalnızca Finlandiya popülasyonunda tanımlanmış olan uzuv-kuşak kas distrofisi tip 2J'ye neden olur. ANO5 genindeki mutasyonlar, uzuv kuşağı kas distrofisi tip 2L'ye neden olur. Diğer birkaç gendeki mutasyonlar, uzuv-kemer musküler distrofi tipleri 2I, 2K, 2M ve 2N dahil olmak üzere, distroglikanopatiler olarak adlandırılan uzuv-kemer kas distrofisi formlarına neden olur.
Ekstremite kuşağı kas distrofisinin diğer nadir biçimleri, bazıları tanımlanmamış olan diğer bazı genlerdeki mutasyonlardan kaynaklanır. Ek olarak, bazı araştırmacılar tarafından kol ve bacak kas distrofisi olarak sınıflandırılan belirli formlar için, diğer araştırmacılar bunları miyofibriler miyopati , Emery-Dreifuss kas distrofisi , dalgalanma kas hastalığı veya Pompe hastalığı gibi farklı, ilgili bozukluklarla gruplandırmayı önermektedir .
KALITIMI
Kol-bacak kas distrofisi farklı kalıtım modellerine sahip olabilir.
Bu durumun çoğu formu , otozomal resesif bir modelde kalıtılır ; bu, her hücredeki genin her iki kopyasının da mutasyona sahip olduğu anlamına gelir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri, mutasyona uğramış genin bir kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını göstermezler.
Bu müsküler distrofinin birkaç nadir formu, otozomal dominant bir modelde kalıtılır; bu, her hücrede değiştirilmiş genin bir kopyasının bozukluğa neden olmak için yeterli olduğu anlamına gelir.
TEDAVİ
Kol-bacak kas distrofisinde kök hücre ve gen tedavisi ile ilgili çalışmalar bulunmaktadır.
KAYNAKLAR
Understanding Neuromuscular Disease Care. IQVIA Institute. Parsippany, NJ. (2018).
Narayanaswami, P. et al. Evidence-based guideline summary?: Diagnosis and treatment of limb-girdle and distal dystrophies. Neurology (2014).
Wicklund, M. P. Limb-Girdle Muscular Dystrophies. in Encyclopedia of the Neurological Sciences (2014). doi:10.1016/B978-0-12-385157-4.00623-0
Menezes, M. P. et al. Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology (2012). doi:10.1212/WNL.0b013e318250d839
Cagliani, R. et al. A CAV3 microdeletion differentially affects skeletal muscle and myocardium. Neurology (2003). doi:10.1212/01.WNL.0000097320.35982.03
Kirschner, J. & Bönnemann, C. G. The Congenital and Limb-Girdle Muscular Dystrophies: Sharpening the Focus, Blurring the Boundaries. Archives of Neurology (2004). doi:10.1001/archneur.61.2.189
Minetti, C. et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. (1998). doi:10.1038/ng0498-365
Hauser, M. A. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum. Mol. Genet. (2002). doi:10.1093/hmg/9.14.2141
Angelini C, Giaretta L, Marozzo R. An update on diagnostic options and considerations in limb-girdle dystrophies. Expert Rev Neurother. 2018 Sep;18(9):693-703. doi: 10.1080/14737175.2018.1508997. Epub 2018 Aug 21. Citation on PubMed
Chu ML, Moran E. The Limb-Girdle Muscular Dystrophies: Is Treatment on the Horizon? Neurotherapeutics. 2018 Oct;15(4):849-862. doi: 10.1007/s13311-018-0648-x. Citation on PubMed or Free article on PubMed Central
Khadilkar SV, Patel BA, Lalkaka JA. Making sense of the clinical spectrum of limb girdle muscular dystrophies. Pract Neurol. 2018 Jun;18(3):201-210. doi: 10.1136/practneurol-2017-001799. Epub 2018 Feb 22. Citation on PubMed
Liewluck T, Milone M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018 Aug;58(2):167-177. doi: 10.1002/mus.26077. Epub 2018 Feb 7. Citation on PubMed
Liu W, Pajusalu S, Lake NJ, Zhou G, Ioannidis N, Mittal P, Johnson NE, Weihl CC, Williams BA, Albrecht DE, Rufibach LE, Lek M. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet Med. 2019 Nov;21(11):2512-2520. doi: 10.1038/s41436-019-0544-8. Epub 2019 May 20. Citation on PubMed
Mah JK, Korngut L, Fiest KM, Dykeman J, Day LJ, Pringsheim T, Jette N. A Systematic Review and Meta-analysis on the Epidemiology of the Muscular Dystrophies. Can J Neurol Sci. 2016 Jan;43(1):163-77. doi: 10.1017/cjn.2015.311. Citation on PubMed
Mitsuhashi S, Kang PB. Update on the genetics of limb girdle muscular dystrophy. Semin Pediatr Neurol. 2012 Dec;19(4):211-8. doi: 10.1016/j.spen.2012.09.008. Citation on PubMed
Straub V, Murphy A, Udd B; LGMD workshop study group. 229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017. Neuromuscul Disord. 2018 Aug;28(8):702-710. doi: 10.1016/j.nmd.2018.05.007. Epub 2018 May 24. No abstract available. Citation on PubMed
Thompson R, Straub V. Limb-girdle muscular dystrophies - international collaborations for translational research. Nat Rev Neurol. 2016 May;12(5):294-309. doi: 10.1038/nrneurol.2016.35. Epub 2016 Apr 1. Citation on PubMed
Wicklund MP, Kissel JT. The limb-girdle muscular dystrophies. Neurol Clin. 2014 Aug;32(3):729-49, ix. doi: 10.1016/j.ncl.2014.04.005. Citation on PubMed.
https://medlineplus.gov/genetics/condition/limb-girdle-muscular-dystrophy/#referenc
https://www.mda.org/disease/limb-girdle-muscular-dystrophy
- Marie Curie Nobel Ödülüne Giden Yol
- Zona Aşısı Kalp Hastalığı Riskini Azaltıyor
- Waardenburg Sendromu Genetiği
- Prader-Willi Sendromu (PWS) Genetiği
- Cornelia de Lange Sendromu (CdLS) Genetiği
- Tıp Fakültesi Dekanı Prof. Dr. Haydar BAĞIŞ 'ın Beyaz Önlük Giyme Töreni Açılış Konuşması
- X'e Bağlı Öldürücü İnfantil SMA-2 (X'e Bağlı Spinal Müsküler Atrofi-2)
- Fragile X İlişkili Primer Over Yetmezliği (FXPOI) Genetiği (Erken yumurtalık yetmezliği)
- Ailesel Hiperkolesterolemi Genetik Yaklaşım
- Kalıtsal Polikistik Böbrek Hastalığına Genetik Yaklaşım
- Sağlıklı Uzun Ömürde Epigenetik Hafızanın Organ Yaşınızdaki Rolü
- Friedreich Ataksisi Genetiği
- Prof. Dr. Bağış: Trafik Kazası Dışındaki Her Hastalık Genetiktir
- Şizofreninin Genetik Yönü Varmıdır ?????
- Waardenburg Sendromu Genetiği
- Göz Renginde Genetiğin Rolü Nedir ?
- Noonan Sendromuna Genetik Yaklaşım.
- Akondroplazi (Cücelik) Genetiği
- Hipohidrotik Ektodermal Displazi Genetiği
- Kistik Fibrosiz Genetiği ve Tedavisinde En Son Gelişmeler
- Adrenolökodistrofi (X-ALD) ( Lorenzo’nun Yağı)
- Hemofili Genetiğinde Gen Tedavisi
- Prof.Dr.Haydar Bağış’ın İsmi Doğum Yeri BESNİ'de Bir Caddeye Verildi.
- Li-Fraumeni Sendromu (LFS) Genetiği
- Cowden Sendromu (PTEN Hamartom Tümör Sendromu)
- Limb-Girdle Musküler Distrofi (LGMBD)( Kol-Bacak Kas Distrofisi) Genetiği
- EPİGENOM VE EPİGENETİK NEDİR
- Mesane Kanseri Genetiği
- SHOX Geni ve İdiyopatik Boy Kısalığı
- Alfa-Talasemi Mental Retardasyon Sendromu
- Albino (Albinizm) Hastalığı Genetiği
- Kromozom 10q26 delesyon sendromlu hastalar
- MTHFR Geni Hakkında Bilmeniz Gerekenler
- Ailevi Akdeniz Ateşi (FMF) Genetik Geçişli Hastalıktır
- Faktör V Leiden Mutasyonu ve Derin Ven Trombozu
- BRCA1/2 Genleri Meme Over Kanseri
- Nörofibromatosiz Tip 1 (NF1) Genetiği
- NADİR GENETİK HASTALIKLAR
- Genetik Yatkınlık Nedir ?
- D-dimer testi ve COVID-19
- Kulaklarda Franks işaretinin akut aort diseksiyon bağlantısı var mı?
- COVID-19 Aşıları Erkeklerde İktidarsızlık ve Kısırlık Yapmaz.
- MTHFR C677T, Faktör V Leiden, PAI 4G/5G, Protrombin G20210A Mutasyonu
- Geleceğin 100 Mesleği ve YKS Tercihi
- İki Doz COVID-19 Aşılı Kişilere CDC Önerileri
- COVID-19 mRNA Aşıları Hakkında Merak Etikleriniz
- COVID-19 Virüsünün Varyantları ve Aşılar
- COVID-19 Yeni Nesil Aşılar Nasıl Çalışır?
- Pıhtılaşma Bozukluğunda Factor V Leiden (FVL) Trombofili Genetiğinin Rolü
- Gençlerde Erişkin Tip Diyabette (MODY) Genetiğin Rolü
- Otizimin Genetik ve Çocukluk Aşılarıyla Bağlantısı Varmı?
- Genomik ve Genetik
- DOWN SENDROMLU HER ÇOCUĞUMUZ KESİNLİKLE KROMOZOM ANALİZİ YAPTIRMALI
- Ailesel Kanserler (Li-Fraumeni Sendromu ) Genetiği
- Doğuştan Kalp Hastalığının Genetik Yönü Varmıdır?
- KÖPEKLER COVID-19 KOKLAYARAK TESPİT ETMEYE BAŞLADI
- Tourette Sendromunda Genetiğin Rolü
- COVID-19 ile Karıştırılan Üst ve Alt Solunum Yolu Enfeksiyon Hastalıkları
- Renin – Anjiyotensin – Aldosteron Sistem Blokerleri ve Covid Riski Arasında İlişki Araştırıldı
- Omurilik Felcinde (Parapleji) Kök Hücre Yaklaşımı
- Genetik Hastalıkların Çocuklarıma Geçme Olasılığı Varmıdır?
- ADYÜ Tıbbi Genetik ve Tıbbi Genetik Uzmanlığı Eğitimi
- Geleceğin Ramboları Genom Düzenleme İle Yaratılmak mı İsteniyor?
- ÇOCUKLUK ÇAĞI AŞILARI HAKKINDA BİLENDE KONUŞUYOR BİLMEYENDE......
- Sağlığınızın Yolu Hissettiğiniz Genç Yaşa Bağlı......
- Adıyaman Üniversitesi Deney Hayvanları Sertifika Kurs Kayıtları Başlamıştır.
- En Popüler 65 Sosyal Medya Siteleri Hakkında Daha Fazla Bilgi Edinin
- 21 nci Yüzyılda Doktorları Neler Bekliyor ve Bakın Onlar Ne ile Uğraşıyorlar...........
- GENETİK DANIŞMANLIK HANGİ DURUMLARDA ALINMALI
- YÖK TARAFINDAN ARAŞTIRMA VE ADAY ARAŞTIRMA ÜNİVERSİTELERİNİN İKİ YILLIK PERFORMANSLARI AÇIKLANDI
- Nobel Fizyoloji ve Tıp 2019 Ödülünün Arka Planı
- Parkinson Hastalığının Genetik Yönü Varmıdır?
- Karaciğer Transplantasyonu Yerine Yeni Bir Kök Hücrenin Nakli.
- Adıyaman Üniversitesi (ADYÜ) Kimliği
- DOKTOR ADAYLARINI ADIYAMAN ÜNV. TIP FAK. BEKLİYORUZ.
- Dikkat eksikliği hiperaktivite bozukluğunun genetik bağlantısı?
- Erkekler Dikkat Ereksiyon Bozukluğunun Genetik Bağlantısı Ortaya Kondu.
- Şekerli içecekler ve kanser arasında olası bir bağlantı ortaya kondu
- Çölyak Hastalığının Belirtileri Nelerdir
- GENOM DÜZENLEME TEKNOLOJİSİ İLE OTİSTİK MAYMUN MODELİ
- OTİZİM ve BURUN İÇİ OKSİTOSİN UYGALAMASININ FAYDA VE ZARARLARI
- MUTLULUĞUNUZ İÇİN DOPAMİNİNİZİ NASIL ARTTIRILABİLİR.SİNİZ
- Mizacımız Genetik Kodlarımız Tarafından Belirleniyor........
- Rett Sendromu Otizm ya da Down Sendromu ile Karıştırılan bir Hastalık.
- Genetik Test Nedir?
- Eğitimde STEM Yaklaşımının Önemi.
- Bilim Adamları Y Kromozomsuz Erkek Fareler Yetiştirdiklerini Açıkladılar.
- 21 Mart Dünya Down Sendromu Günü ve Down Sendromu Nedir?
- KANSER İMMÜNOTERAPİSİ NEDİR?
- Dünyanın ilk genetik tasarımlı bebekleri Çin'de doğdu
- PARKİNSON HASTALIĞI İLE APANDİS ARASINDA İLİŞKİ BULUNDU.
- CRISPR köpeklerde Duchenne musküler distrofi ilerlemesini durdurdu.
- BAKIR ve ALZHEİMER HASTALIĞI
- Doymuş Yağ Kullanımında Bilinen Yanlışlar........
- OTİZMLE HAMİLE ANNENİN BAĞIRSAK MİKROBİYOMU ARASINDAKİ İLİŞKİ
- KANSERDE SIĞIR KIKIRDAĞI KULLANIMININ ÇOK ETİKİLİ OLDUĞU ORTAYA KONDU.
- ALKALİ SU TÜKET DAHA SAĞLIKLI YAŞA..........
- Dünyanın İlk Klon Maymunları Zhong Zhong ve Hua Hua 7 aylık oldu.
- Amerikan Ulusal Kanser ve Sağlık Enstitüleri (NIH) Hakkında Bilgi
- GENOMDAKİ TRANSPOZONLAR BENCİL PARAZİTLERMİ ?
- Sigara İçen Y kromozomunu yitiren erkeklerin, yitirmeyenlerden ortalamada 5.5 yıl daha erken öldüğü tespit edilmiş.
- FDA, Kolorektal Kanser, Lösemi ve Melanom için Yeni Tedavileri Onayladı
- BAĞIRSAK MİKROPLARININ OBEZİYETE ETKİSİ
- GENOM MÜHENDİSLİĞİNDE CRISPR/CAS9 TEKNOLOJİSİNİN DENEYSEL HAYVAN MODELLERİNDEKİ YERİ
- GEBZE TEKNİK ÜNİVERSİTESİ 2. GENETİK GÜNLERİ ETKİNLİĞİ
- 28 Şubat Nadir Hastalıklar Günü. Nadir Görülen Hastalıkların% 80'i Genetik Kökenlidir
- MEME KANSERİNİN YAYILIMINDA BİRÇOK GIDADA BULUNAN ASPARAJİNİN SORUMLU OLDUĞU TESPİT EDİLDİ.
- Kolon Kanserinde Bağırsak Bakterisinin Rolü Olduğu Anlaşıldı.
- Yeni Otizm İlaçları, Klinik Öncesi Çalışmada Umut Vaad Etti
- OTİSTİK ÇOCUKLARA GAİTA MİKROBİYOMUNUN TRANSFERİ
- ERKEKLERDE FAZLA VİTAMİN B6 VE B12 AKCİĞER KANSER RİSKİNİ ARTTIRABİLİR DİKKAT...
- CRH geninde keşfedilen epigenetik değişiklikler, intihar riskini artırıyor....
- FDA, 2. Gen Terapisini Onayladı
- Kolesterol testleri için emeklilik zamanı geldi mi?
- Her 30 Dakikada Bir Hareket Et ÖLÜM RİSKİNİ AZALT.............
- Homeobox Genleri Nelerdir......
- Genom düzenleme ve CRISPR-Cas9 nedir?
- Kalp krizi ve felç gibi şeylere karşı riskinizi öğrenmek istermisiniz.
- Cebinizdeki Çantanızdaki Akıllı Telefonunun Zararları Nelerdir?
- Tütün Yanarken Oluşan Yüzlerce Farklı Element Epigenetik Modifikasyonlara Yol Açıyor
- 2017 NOBEL TIP FİZYOLOJİ ÖDÜLÜ SİRKADİYEN RİTMİNİ KONTROL EDEN GENLERE VERİLDİ
- Diyabet İlacı Metformin Bazı Kanser Türlerine Karşı
- SMA Hastalarında Yeni ve Pahalı Tedavi Dönemi Başladı
- TÜRKİYE'de NASIL DOÇENT OLUNUR.
- Bir Günde 2 Yumurta Yemeye Başlarsanız Vücudunuza Olabilecek 9 Olumlu Şey.
- Bağırsamızdaki Mikroplar Yeni Organımız mı?
- BEBEĞİN OBEZİTE RİSKİ ANNENİN BESLENMESİNE BAĞLI OLARAK RAHİMDE BAŞLIYOR (GENLERİN AÇILIP KAPANMASINDAN BAŞLIYOR)
- Çay tüketimi kadınlarda epigenetik değişikliklere neden olduğu ortaya kondu
- İSTANBUL BESNİLİLER DAYANIŞMA DERNEĞİNDE AKRABA EVLİLİKLERİNDE GENETİK YAKLAŞIM KONFERANSI
- NATURE GENETİK DERGİSİ 25 YILDAKİ EN İYİ MAKALELERİNİ SEÇTİ
- Çölyak Hastalığında Reovirusların Rolü Ortaya Kondu
- cjCRISPR/Cas9 ile KÖRLÜK TEDAVİSİ
- CRISPR-Cas9 İLE NADİR KAN HASTALIKLARININ TEDAVİSİNDE YENİ UMUT
- NADİR GÖRÜLEN GENETİK HASTALIKLARIN TEŞHİSİNDE YÜZ TANIMA YAZILIMI
- Öğretmeni Şok Eden Cevap. Öğretmen Öğrenciye Sorar Ne Olmak İstiyorsun? Öğrenci Haydar BAĞIŞ Olmak İstiyorum Diye Cevap Verir
- UYKU YOKSUNLUĞU KALP HASTALIKLARINA DAVETİYE ÇIKARIYOR DİKKAT...........
- 8 Mart Dünya Kadınlar Gününüz Kutlu Olsun
- Etnisite Gibi Kültürel Farklılıklar, DNA Üzerinde İz Bırakabilir
- ZAMANINDA UYKU BEYNİ SIFIRLAYIP HAFIZAYI GÜÇLENDİRİP HASTALIKLARA DİRENÇLİ OLMAMIZI SAĞLIYOR.
- İnsanda Körelmiş 10 Organ
- 2016 Yılındaki En İyi 10 Tıbbi Yenilik
- FAZLA KİLOLARINIZ DNA METİLASYONUNU ETKİLER
- 10 Ocak Çalışan Gazetecilerimizin Gününü Kutlarım
- IPS KÖK HÜCRELERİ DONÖRÜN YAŞININ GENOMİK İZLERİNİ TAŞIYOR
- ÇOCUK İSTİSMARI İNTİHAR ve EPİGENETİK
- AKRABA EVLİLİĞİ GENETİK DANIŞMANLIK.........
- TIBBİ GENETİK VE KLİNİK UYGULAMALARI KİTABIMIZ RAFLARDA
- Periyodik Tablo’ya Dört Yeni Element Resmen Eklendi
- YENİ YILINIZ KUTLU VE MUTLU OLSUN
- EMBRİYONUN GENOM BÜTÜNLÜĞÜNÜN NASIL KORUNDUĞU KEŞFEDİLDİ.
- BAĞIRSAK MİKROBİYOMUNA DİKKAT YOKSA PARKİNSON OLABİLİRSİNİZ..........
- Kediler Hakkında Bilmediğimiz 10 şey
- Akciğer Kanserli Hastada İlk Defa CRISPR-Cas9 Tekniği ile Düzeltilmiş Hücre Nakli.....
- Doçentlik Başvurusu Yapan Adayların Dikkatine?
- CRISPR/Cas9 İLE SAĞLIKLI İNSAN EMBRİYO ÜRETİLMESİ
- GEBELERDE SABAH BULANTISI "'Düşük Riskini Azaltıyor'
- BAZI AĞRI KESİCİLER BAZI KİŞİLERDE KALP YETMEZLİĞİ RİSKİNİ ARTTIRABİLİR.
- SİGARA İÇENLERİN AKCİĞERİNDE 150 MUTASYON SAPTANDI.....
- Yeni bir tüp bebek yöntemiyle genetiği değiştirilmiş sağlam bir bebek dünyaya geldi
- Panorama Test - Non İnvaziv Prenatal Test (NIPT)
- GENETİKTE SIK SORULAN SORULAR
- Doğacak bebekte kalıtsal hastalık görülme riskinin öngörülmesi amacıyla yapılan genetik tarama testi nedir.
- Prof.Dr.Haydar BAĞIŞ Nadir Hastalıklar Konusunda Açıklama Yaptı.
- Genetik Uzmanı Prof. Dr. Haydar Bağış, insanların yedikleri ve düşündüklerinin genlerin çalışmasını etkilediğini kaydetti
- B Vitaminlerinin Beynin Yaşlanması Üzerine Etkileri
- ADYÜ TIP FAK. ÖĞRENCİLERİ SAĞLIK PROJESİ SUNUMLARI
- Prof.Dr.HAYDAR BAĞIŞ HABERTÜRK TV ÖTEKİ GÜNDEM PROGRAMINA CANLI YAYIN KONUĞU OLACAK
- Klonlama Teknolojisi, Türkiye İçin Yeni Bir Gelir Kapısı Olabilir
- Bilim depresyonu alt eden besteyi buldu?
- AKADEMİK TEŞVİK ÖDENEĞİ YÖNETMELİĞİ YAYINLANDI
- SIÇRAYAN GENLER BENCİL Mİ?
- Hücrenin Sayı Sayma Kabiliyeti
- EHLİYET, LİYAKAT VE ÜNİVERSİTELER……
- 2014 Yılı Ar-Ge Faaliyetleri Anketi Sonuçları
- İnsan Bedeni Hakkında Şaşırtıcı Gerçekler
- Türkiye'deki Klon Sığırlar Çoğalmaya Devam Ediyor.
- Türk Bilim Adamı Aziz Sancar Hocamız Bu yıl NOBEL Kimya Ödülüne Layık Görüldü.
- Bilim adamları Petri kabında kök hücreden minyatür insan böbreği üretmeyi başardı.
- İnsanı Felakete Götüren 10 Şey
- Klonlanmış hayvanın eti zararlı mı?
- İnsan embriyosunda genetik değişimler yapılıp, DNA'mız ile mi Oynanacak Acaba ?
- Meme Kanserine Genetik Yaklaşım
- Baştan Sona Avrupa Turunda Gezdiğimiz Ülke ve Şehirler
- KAMAG 1007 Programı Kapsamında Yükseköğretim Kurulu Başkanlığı Adına Yeni Çağrılar Açıldı
- BABALIK TESTİ (PATERNİTE) NEDİR VE NASIL YAPILIR
- TIBBİ GENETİKTE KARDİYOVASKÜLER RİSK PANELİ NEDİR?
- Prof. Dr. Haydar BAĞIŞ İnsanı Klonlamak Hayvanı Klonlamaktan Daha Kolay Diyor.
- YÖK Hastaneleri yok saydı!
- TÜBİTAK Doktoralı Kişilerin İstihdamını Teşvik Eden Yeni Bir Çağrı Açtı
- URAP: ODTÜ Enformatik Enstitüsü Yayın Değelendirme Kriterleri
- Akademisyenlerin ‘atıf’ çetesi
- Adıyaman Fen Lisesi Öğrencileri Tıp Fakültemizi ve Beni Ziyarete Geldiler
- Prof.Dr. Haydar BAĞIŞ diyor ki bebeğin cinsiyetini erkek belirliyor Toplumda sıkça kullanılarak yer edinen 'erkek adamın erkek çocuğu olur' düşüncesinin yanlış olduğu ortaya çıktı. 24.03.2011
- Prof.Dr. Haydar BAĞIŞ'ın da Bölüm yazarlığı yaptığı "Current Aplications of Biotechnology" kitabı yayınlandı.
- ÜNLÜ TİYATRO SANATÇIMIZ ZEKİ ALASYA YI KAYBETTİK
- YÖK Başkanı Prof.Dr. Yekta SARAÇ 'Üniversiteler susturulmuş' değil isteksiz
- Rektörlük Seçimleri ve Muhalefet
- Prof.Dr.Haydar BAĞIŞ'ın Görüşü YENİ YOK YASASI
- Adıyaman Üniversitesi Rektörlük Seçim Sonuçları
- REKTÖRLÜK SEÇİMİNİ BİLİMİN KAZANMASI DİLEĞİYLE
- Tüm Hocalarımıza Teşekkür Ederim
- HEDEFİMİZ; SÖZÜ DİNLENEN, DANIŞILAN, BİLGİ ÜRETEN, ULUSLARARASI SAYGINLIĞI OLAN BİR ÜNİVERSİTE OLUŞTURMAKTIR. BUNU HEP BİRLİKTE BAŞARACAĞIZ VE ÜNİVERSİTEMİZİ ORTAK AKILLA BERABER YÖNETECEĞİZ.
- Prof.Dr.Haydar BAĞIŞ REKTÖR ADAYLIĞI BAŞVURU DİLEKÇE VE FORMLARINI REKTÖRLÜĞE TESLİM ETTİ.
- Kalıtsal Hastalıklardan Temizlenmiş Tüp Bebekler
- Prof.Dr. Haydar BAĞIŞ Kitapları
- Prof. Dr. Bağış, Seçim Çalışmalarının Yanında Bilimsel Çalışmalarına Devam Ediyor
- Prof.Dr.Haydar BAĞIŞ'ın Projesi TÜBİTAK Akademik Başarı Öyküleri Kitabı’ nda Yer Aldı
- Hastalığım Süresince Arayan Tüm Dostlarıma Teşekkür Ederim.
- Tıp Eğitiminde E-Öğrenme
- Adaylara değil, partilere, liderlere oy verilecek
- Prof.Dr.Haydar BAĞIŞ SANKO Üniversitesi'nde "Moleküler Tıpta Biyomühendislik ve İnovasyon" konulu sempozyumunda konuştu..
- YDS SINAV STRATEJİSİ CÜNEYT BADEMCİ
- ÜNLÜ AKADEMİSYEN PROF. DR. HAYDAR BAĞIŞ KMÜ’LÜ ÖĞRENCİLERLE BULUŞTU
- Berber Çıraklığı Yaptım Ustamı Fatih semtinde Ziyaret Ettim.
- 14 Mart Tıp Bayramı Kutlaması.
- 8 MART DÜNYA KADINLAR GÜNÜ MESAJI
- "MOLEKÜLER TIPTA BİYOMÜHENDİSLİK VE İNOVASYON" SEMPOZYUMU
- İki kız evladı babası olarak erkek olmaktan utanç duyuyorum. !
- ADYÜ Tıp Fak. Tıbbi Genetik Anabilim Dalına Bağlı Genetik Tanı Biriminde Yılda 2500 Hastaya Genetik Test Hizmeti Verilmektedir.
- Bebeğin cinsiyetini erkek belirliyor
- Prof.Dr. Haydar BAĞIŞ ADYÜ Tıp Fak. Konferans Salonunda "Bilimsel Makale Nasıl Yazılır ve Yayınlanır" Başlıklı Seminer Verdi.
- Adıyaman Ünv. Tıp Fak. Öğrenci Sempozyumu Yaptı.
- Prof.Dr. Haydar BAĞIŞ Başkanlığındaki TÜBİTAK Ekibi Dünyada ilkez "Türk Malı Buzul Ayısı" ismini verdiği Donmaya Dirençli Transgenik Fare Soyları Geliştirildi
- Prof. Bağiş, OMÜ’de Transgenik ve Klon Hayvan Üretimini anlattı
- 8. Moleküler Biyoteknoloji Bahar Okulu, 19-22 Nisan, 2013
- Prof.Dr. Haydar BAĞIŞ Başkanlığındaki Ekip tarafından Ülkemizde İlk Defa Transgenik Zebra Balığı Yapıldı.
- Prof.Dr. Haydar BAĞIŞ'ın projesi TÜBİTAK Akademik Başarı Öyküleri Kitabı’ nda Yer aldı.
- Tıp Fakültesi Önünde Basın Bildirisi - Adıyaman 16 Eylül 2009 Çarşamba 15:03
- Prof. Dr. Haydar Bağış: Transgenik hayvan çalışmalarının bilime katkıları isimli sunum yaptı.
- Fare sütü kansere çare olacak. Donmaya dirençli, “Türk malı buzul ayısı’’ adı verilen bir fare geliştirerek adlarını duyuran TÜBİTAK araştırmacıları bir başarıya daha imza attı
- KLONLAMAYA TEPKİLER
- TÜBİTAK'tan müthiş bir proje daha TÜBİTAK, fare sütünde, özellikle kanser tedavisinde kullanılan insana ait 'interferon gamma' isimli bir protein üretti
- TÜBİTAK interferon gamma üretti
- Prof.Dr. Haydar BAĞIŞ ve Prof.Dr. Munis DÜNDAR Editör ve yazarlığını yaptığı " Modern Biyoteknoloji Uygulamaları" Başlıklı Kitap
- Konu: EŞCİNSELLİK GENETİK Mİ? PELİN ÇİFT: Prof Dr Haydar BAĞIŞ ve Prof Dr Uğur ÖZBEK-